Note: These tutorials are incomplete. More complete versions are being made available for our members. Sign up for free.

Smith-Waterman Algorithm
In the last section, we presented the mapping problem at three levels of difficulties. A computational solution encompassing all three levels exists and was proposed long back in 1981 by Temple F. Smith and Michael S. Waterman. Smith-Waterman algorithm performs local alignment to find the best match of a short string within a large reference sequence. It uses dynamic programming approach and is the local implementation of a similar global search algorithm proposed by Needleman and Wunsch.
Rationale behind the Algorithm
Instead of going straight to the algorithm, let us first explain the rationale behind it so that you may derive it yourself without our help. Moreover, understanding the rationale will help you understand how BLAST, FASTA and other fast and heuristic implementation of Smith-Waterman, speed up the native algorithm. BLAST and FASTA will be presented in Section 4.
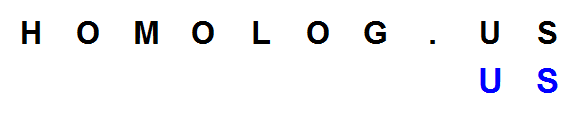
Suppose you want to instruct the computer to find ‘US’ in ‘HOMOLOG.US’. How would you design your code? One way is to find all hits to the first letter U in the reference sequence and extend from there. Once you locate all Us, check whether the next letter is S and so on.
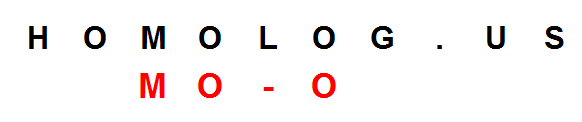
The extension is easy for perfect matches like ‘US’, but more difficult for words like ‘MOO’ with gaps or with imperfect matches. After the first M is found, the algorithm needs to allow for the possibilities of MO and M_. In the next step, it needs to keep count of MOO, MO_, M__ and M_O and so on. The number of possibilities increase exponentially with longer sequences. How does one take care of that?
The first inclination to keep the number of possibilities under control is to hard-code the maximum number of allowed gaps to 1, 2 or 3. That is not optimal, because longer sequences may have more insertions and deletions. A better approach is to assign a positive score to each perfect match and a negative score to each gap. When a substantial number of gaps accumulate, total score drops to zero, thus leading to rejection of sub-word under consideration.
In a further refinement, a large positive score can be assigned to perfect matches and small positive score for some imperfect matches, such as for similar amino acids in case of protein searches.
Detailed Implementation