
Qudaich - a Smart Sequence Aligner
Readers may find a new paper titled ‘Qudaich: a Smart Sequence Aligner’ interesting. Their unusual and effective way of building Suffix array may be useful for other research problems.
Problem it solves
The authors developed an algorithm to make rapid search for nucleotide or protein sequences within a database. Do we need yet another search program? Well, it may be needed in the problem space the authors are working on, which is searching for genes or proteins in a database.
Benchmarks
The benchmarks can be seen from Tables 3 and 4 of the paper.
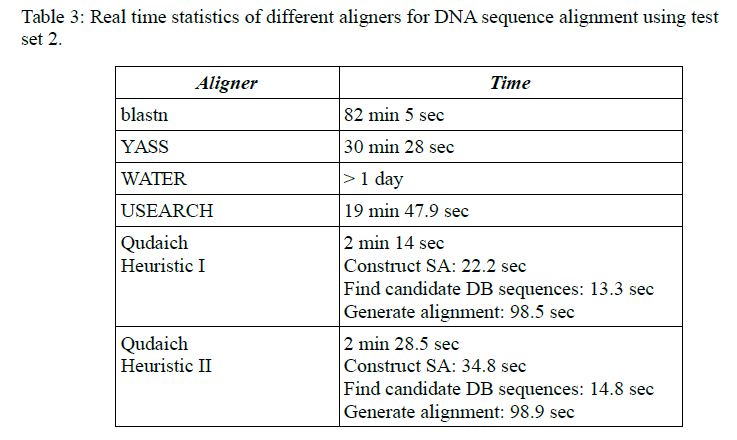
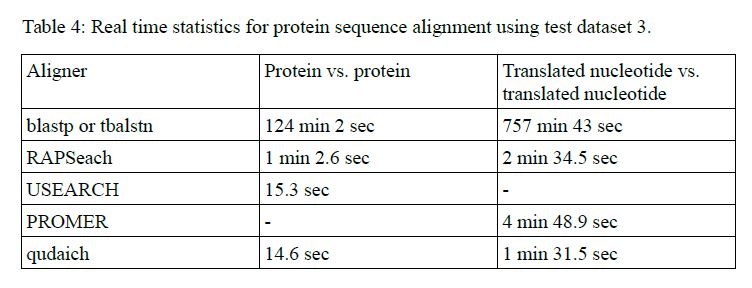
Algorithm
The main algorithmic novelty of Qudaich is in building a single suffix array using both database and query sequences. All other algorithms use only the database and does not include query.
Inclusion of query sequences allows the suffix array patterns to coalesce around the sequences within the query. That helps the authors use heuristic methods to identify the database entity, where the query is likely to be present. After finding the database entity, Qudaich performs full Smith-Waterman-Gotoh alignment within the database.
Qudaich uses a novel algorithmic structure to look for the candidate database sequences efficiently. Most aligners keep the database and query sequences separate, but in qudaich both database and query sequences are organized together. This novel organization has two main advantages. It accelerates the searching of candidate database sequences by clustering several query sequences; and, it allows us to construct powerful heuristics to limit the search space. However, the primary disadvantage of using the query and database sequences together is that the sequence indices need to be rebuilt for each different comparison.
More details here -
Qudaich uses a suffix array as the primary data structure. It constructs a single suffix array using all database and query sequences.
In our algorithm, a long string, S is constructed by concatenating all the database sequences, the reverse complement of the database sequences and then all the query sequences. Then using S, a suffix array, SA is constructed.
** Abstract:** Next generation sequencing (NGS) technology produces massive amounts of data in a reasonable time and low cost. Analyzing and annotating these data requires sequence alignments to compare them with genes, proteins and genomes in different databases. Sequence alignment is the first step in metagenomics analysis, and pairwise comparisons of sequence reads provide a measure of similarity between environments. Most of the current aligners focus on aligning NGS datasets against long reference sequences rather than comparing between datasets. As the number of metagenomes and other genomic data increases each year, there is a demand for more sophisticated, faster sequence alignment algorithms. Here, we introduce a novel sequence aligner, Qudaich, which can efficiently process large volumes of data and is suited to de novo comparisons of next generation reads datasets. Qudaich can handle both DNA and protein sequences and attempts to provide the best possible alignment for each query sequence. Qudaich can produce more useful alignments quicker than other contemporary alignment algorithms.