
New Paper on Comparing Genome Assemblies
How to compare genome assemblies coming out of various programs and tell which one is correct?
De novo likelihood-based measures for comparing genome assemblies
We demonstrate that our de novo score can be computed quickly and accurately in a practical setting even for large datasets, by estimating the score from a relatively small sample of the reads. To demonstrate the bene?ts of our score, we measure the quality of the assemblies generated in the GAGE and Assemblathon 1 assembly bake-offs with our metric. Even without knowledge of the true reference sequence, our de novo metric closely matches the reference based evaluation metrics used in the studies and outperforms other de novo metrics traditionally used to measure assembly quality (such as N50). Finally, we highlight the application of our score to optimize assembly parameters used in genome assemblers, which enables better assemblies to be produced, even without prior knowledge of the genome being assembled.
The paper presents a probabilistic model of genome assembly to compute ‘likelihood of an assembly’. True assembly is expected to obtain the maximum likelihood.
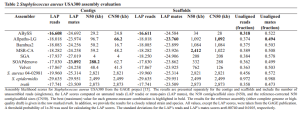
To demonstrate the approach, the paper three tables like above comparing various assemblers, both at the contig and scaffold levels and telling you which is closest to the ‘truth’. Two are for bacterial genomes and one for human. For some strange reason, they do not include SPAdes.
Conclusion
Likelihood-based measures, such as ours proposed here, will become the new standard for de novo assembly evaluation.