New Nanopore Paper by Phil Ashton
Readers of our blog are familiar with Philip Ashton’s excellent work (see here and here). He recently published a new paper on nanopore sequencing that you may enjoy. In twitter, it is being presented as the ‘first serious analysis of Nanopore data’.
Short-read, high-throughput sequencing technology cannot identify the chromosomal position of repetitive insertion sequences that typically flank horizontally acquired genes such as bacterial virulence genes and antibiotic resistance genes. The MinION nanopore sequencer can produce long sequencing reads on a device similar in size to a USB memory stick. Here we apply a MinION sequencer to resolve the structure and chromosomal insertion site of a composite antibiotic resistance island in Salmonella Typhi Haplotype 58. Nanopore sequencing data from a single 18-h run was used to create a scaffold for an assembly generated from short-read Illumina data. Our results demonstrate the potential of the MinION device in clinical laboratories to fully characterize the epidemic spread of bacterial pathogens.
The paper has two interleaved stories - one on Oxford nanopore technology and one on genome modification in bacteria leading to antibiotic resistance. We will cover the MinION part here and elaborate on the biological and medical aspects of antibiotic-resistant bacteria in a later blog post.
Speaking of nanopore, the most important contribution of the paper is its estimation of error profile. We will mention several key observations in the following sections.
1. Use only 2D reads in the future
MinION generates three types of reads - template, complement and 2D. Among them, 2D reads use data from two measurements of the same read to reduce the error rate. Based on our understanding of the paper, it will be safe to ignore the first two categories due to low accuracy and use only the 2D reads.
2. Accuracy is 95%, 85%, 10%, 72%
When it comes to the accuracy of Oxford nanopore reads, we have seen all kinds of numbers in the past, ranging from Mikheyev’s 10% to 95%. If this controversy appears too similar to ENCODE’s estimate of functionality of human genome, that is not a coincidence, given that Ewan Birney advises both organizations.
The authors have spent good deal of time on this issue and arrived at ~72% for 2D reads. Interestingly, instrument’s own Phred estimates for the same reads are too high at 84.6% and should be revised. What does not make sense is why accuracy improves to only 72% for 2D from ~62% for ‘1D’ reads. A simple probabilistic calculation would have given 1-(.38^2) or 85% accuracy for 2D. Our naive explanation is that a large fraction of ‘1D’ reads does not get mapped, whereas the mappability improves after 2D correction.
3. Use LAST aligner, not BLAST or BWAMEM
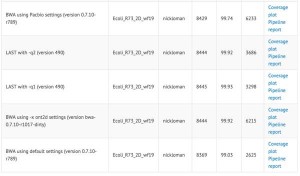
Authors reported LAST aligner working well for ONT reads, but not BLAST or BWAMEM aligners. Why BWAMEM did not work is not clear to us given that it has a setting of ‘-x ont2d’ pegging accuracy at around 65% (corrected, see below). Have no fear - all alignment-related matters are being looked into by Ewan Birney, whose PhD thesis was on ‘Sequence alignment in bioinformatics’. We will update, as soon as we hear from him.
Correction.
Heng Li mentioned that he added ‘-x ont2d’ option in bwa-0.7.11, whereas it was not available in bwa-0.7.10 used by Ashton.
With ‘-x ont2d’, the BWAMEM alignements are as good as LAST for most cases (with even identical alignment scores) except for a tiny fraction of reads.
4. Detailed error profile
The section on detailed error profile is quite informative. Indels are the most common errors just like in Pacbio, and it appears that ONT has difficulty differentiating Gs and Cs in the homopolymer regions. The entire section is presented below.
The error profile of the MinION H125160566 data set was characterized. In the 70.8 megabases of aligned nanopore sequence an indel occurred every 5.9 bases on average. Two-thirds of these indels were deletions (8.6 Mbp), whereas one-third were insertions (4.15 Mbp). The mean deletion length was 1.7 bp, whereas the mean insertion length was 1.6 bp. Both insertion and deletion lengths had a negative exponential distribution (Supplementary Figs. 3 and 4).
A z-score was calculated to identify k-mers that were deleted from mapped MinION reads at a higher-than-expected frequency. When z-scores for deletions of 36 bp were binned by GC content, the tendency of the MinION to skip k-mers containing As and Ts only or Gs and Cs only could be seen (Fig. 1). In particular, there were two 3-mers with a z-score > 3, GGG (z = 3.3) and CCC (z = 3.2), two 4-mers with a z-score > 3, GGGG (z = 4.4) and CCCC (z = 4), and four 5-mers with a z-score > 4, AAAAA, TTTTT, CCCCC and GGGGG. Two more diverse k-mers that had high z-scores were TAGGCA and TAGGGC, with z-scores of 9.6 and 9.5, respectively. When insertions were examined there were two 3-mers with a z-score > 3, CCC (z = 3.7) and GGG (z = 3.6), and two 4-mers with a z-score > 3, CCCC (z = 6.1) and GGGG (z = 6.0).
To identify substitutions, all nonconsensus bases were extracted from the MinION alignment to the Illumina assembly. This showed that A to T and T to A substitutions were approximately half as frequent as other substitutions (Supplementary Table 2). These data suggest that MinION currently has difficulty differentiating Gs and Cs, particularly when these bases are present in homopolymeric tracts.
5. Lex Nederbragt?
Lex Nederbragt became untouchable by mocking the company telling
the truth in his blog and did not get an early access to MinION. We tried our
best in our petition to request early access to Minion nanopore for Lex
Nederbragts group that was not
approved by the company. Therefore, it was encouraging to
see him contributing to the ‘first serious Nanopore paper’. From the
acknowledgement section -
We would also like to thank L. Nederbragt for a thorough review and contributions toward the presentation of Figure 1.
6. Biological discovery - was nanopore sequencing useful?
Speaking of biological discovery, it was not clear how much of authors’ finding of the position and structure of a bacterial antibiotic resistance island came from technological help from Nanopore sequencing and how much came from their own ingenuity. They needed Illumina sequencing to get their work done and used nanopore reads to only put the pieces together. The biggest contribution of nanopore is possibly this one -
The assembly of the Illumina data collapsed these two genes into a single copy, which represented a misassembly that confounded analysis and was only resolved using the MinION reads.
Another question - how can one resolve the assembly failures such as hyp and merA mentioned below?
To demonstrate the utility of MinION sequence for hybrid genome assembly, the Illumina and Minion data for the H125160566 strain were assembled using SPAdes18. This method resolved the genome into 34 contigs, with an N50 of 319 kbp, whereas the Illumina-only assembly produced 86 contigs with an N50 of 154 kbp. The SPAdes hybrid assembly confirmed the yidA insertion site but failed to resolve the complete island structure with contig breaks in the hyp and merA genes, presumably due to the low coverage regions in the Illumina data. SPAdes hybrid assembly demonstrated significant improvement over Illumina-only assemblyfurther improvements would be possible given higher-coverage MinION data.
As we see it, the failure was not due to short or long reads, but rather from GC coverage bias of Illumina Nextera sequencing. So, the only way to solve the problem is to go to some bias-free sequencing technology, unless the lab is ready to do PCR and Sanger sequencing on every assembled genomic segment that is suspect.
Overall, this paper presents the current status of Oxford Nanopore sequencing well, and will be valuable to bioinformaticians for its detailed profiling of error distribution.
-———————————————————
Edit.
Nick Loman posted a comparison table (columns - # aligned, % aligned, mean alignment length) of aligning nanopore R7.3 chemistry to E. coli reference. It shows no difference between ‘-x ont2d’ and ‘-x pacbio’ alignment settings, suggesting that the error rates of Pacbio and ONT2D are similar for this new chemistry. Please note that Ashton et al. likely used an earlier chemistry.