
De Novo Transcriptome Assemblers – Oases, Trinity, etc. - II
We present our apologies for not writing for a while, dear reader. By now you realize that thinking and creativity do not come to us naturally, or otherwise we would be writing fictions or peddle specious economic ideas to unassuming public. Speaking of how creative economic ‘science’ can get, here you can watch our beloved e-con Nobel laureate propose starting an war against space aliens to improve US economy.
We are not that adventurous and shall restrict ourselves to the more earthly topic of transcriptome assembly. As we promised earlier, this series will be presented in four parts. Instead of jumping straight to specific assembly programs, we will first provide a broad overview of the changing field of transcriptomics, and explain the importance of transcriptome assemblers.
Larger Perspectives
We have been working on transcriptomics for the major part of last decade and are observing a rapid paradigm change taking place over the last two years. We are not referring to the ability to generate high-quality gene strucures of well-studied organisms with NGS technology, but a complete overhaul in the way transcriptomics research is done. Let me elaborate.
If you read various genome sequencing papers (mosquito, honey bee, sea urchin, rat, rice), you will notice that most of them spent only few paragraphs describing the genome and devoted the rest on the genes. This is understandable, because an overwhelming majority of biologists are primarily interested in the genes and transcriptomes (differential expression of genes in tissues). Genome sequencing has been the easiest way to get to the genes, but if biologists had a simpler way to determine the genes and their expressions without going through the elaborate process of genome assembly and gene prediction, they would have preferred that option. NGS technology offers exactly that option going straight to the genes and transcriptome.
Let me make clear that I do not want to undermine the importance of genome sequence, and start a civil war like the Liliputs fought to decide whether egg should be broken on big end or little end. The genome sequence is extremely valuable to study cis-regulatory regions that control gene expression. However, those topics come only after genes and expressions are known.
The field of transcriptomics changed greatly over the years. Sequencing was expensive in the first generation. Therefore, most of the money was spent on genome sequencing, and a small amount of funds was reserved to sequence genes in the form of ESTs. The coverage of ESTs was usually poor, and therefore scientists relied primarily on computational gene prediction approaches to annotate the genome. For higher eukaryotes, the gene structures were often wrong or incomplete following this approach.
The cost of manufacturing oligonucleotide arrays came down in the next generation, and an array based approach for finding the genes was invented (human, sea urchin, arabidopsis, etc.). This tiling array-based approach provided two advantages. Firstly, the gene structures were empirically based, and not the product of computer prediction. Secondly, the arrays covered the entire genome, whereas the other alternatives like ESTs only gave information for a few genes. However, designing tiling arrays still required a reference genome. So this method could not be applied on an organism until the genome sequencing is completed.
NGS technologies bring us to the third generation of transcriptomics, where we are able to determine the transcriptome of any organism in any tissue without sequencing the genome before hand. This opens up wide variety of previously inaccessible problems to the geneticists.
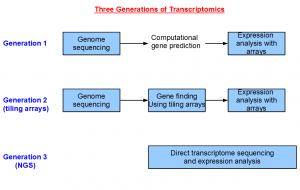
It is possible to do transcriptomics without genomics with NGS technology, because the volume of data is so large that the complete gene structures can be reconstructed at a low cost. You now see clearly how important the transcriptome assemblers in this overall picture.
Difference between transcriptome and genome assemblers
Why do we need separate assembly programs to assemble transcriptomes instead of using de Bruijn-based genome assemblers like Velvet? There are two reasons.
Firstly, as we explained in our article on de Bruijn graph construction, a de Bruijn genome assembler typically assumes that all parts of the de Bruijn graph have uniform number of reads. This is not a good assumption for short read library from a transcriptome, because some genes can be expressed at a very low level and have small number of reads. Some other highly expressed genes will have large number of reads, and they will dominate the assembly. A transcriptome assemblers should be able to adjust for these differences.
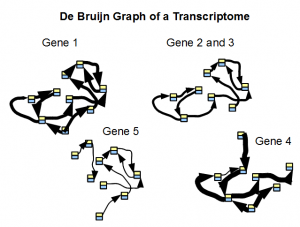
Secondly, the de Bruijn graph of a transcriptome readily separates into small subgraphs of many genes. Transcriptomes are repeat free, and therefore their de Bruijn graphs are not expected to group together unlike de Bruijn graphs from a genome. A good transcriptome assembler should be able to take advantage of this feature.
In the following figure, we show typical structure of de Bruijn graph of a transcriptome as constructed from short reads. Width of arrows connecting nodes represent the number of reads traversing the path.
A similarity between transcriptomics and metagenomics
While on the topic, we point out some similarities between transcriptome assembly and metagenomics. In metagenomics, genetic materials are collected from an environmental sample and all genomes present in the sample are sequenced together. The challenges in assembly of those genomes is not different from transcriptome assembly, because various organisms (and their chromosomes) can be present at different frequencies. Metagenome assembly may pose additional challenges, because the genomes are larger than typical genes and may contain some repeats.
-————–
We have been writing on various topics for a while, but are not sure whether you find them useful. If you do not like to use public comment section, please feel free to email us at ‘samanta at homolog.us’.