
An Elegant Use of NG Sequencing - Finding T-cell Diversity
An year back, we started writing regularly on transcriptomics, next-gen sequencing (NGS) and bioinformatics, starting with two posts - A beginners guide to bioinformatics part I and A beginners guide to bioinformatics part II. Now that our year long effort turned everyone into expert bioinformaticians, we will add a new flavor to our commentaries. Beginning from today, we will also include articles on innovative use of the new technologies with immediate medical or other real- life applications.
Our today’s pick is an immunological application of NGS developed by a research group at Fred Hutchinson Cancer Research Center. They found a neat way to measure T-cell diversity in blood samples through sequencing. After their discovery, the group formed a local company named Adaptive Biotechnologies to help hospitals across the country analyze blood samples. In passing we like to mention that six years back, we tried (and gave up) answering the same question using custom-designed microarrays. The reason of our giving up was primarily financial. When we thought about the experiment, Nimblegen arrays had only 400,000 probes. So, using them to search for millions or billions of potential variable regions in T-cells seemed too expensive.
Let us explain what the Fred Hutch group did starting from basics, because we presume many of our readers do not have background in immunology.
T cells and B cells are two most fascinating components of human immune system. The cells have adapter proteins on their surfaces to detect external invaders. Those adapters change slightly from one T-cell or B-cell to another in the body so that they can collectively detect as many invaders as possible.
T-cells are more complex than B-cells, because they are the ones carrying ‘guns’ to kill invaders. If they get messed up, they can create all kinds of troubles ranging from make you choose wrong girl-friend to attack your own body.
The genetics of T-cells and B-cells is very interesting. All those different adapters come from the same region in the genome. How can one genomic region code for millions of different receptor proteins? The answer is somatic recombination or V(D)J recombination.
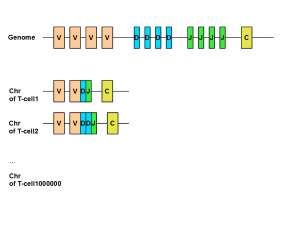
The region of human genome coding for immunoglobulin (B-cell) or T-cell receptor have three types of regions called variable (V), diversity (D) and junction (J). Those V, J and D regions are composed of many small and identical units. When a new T-cell is created from thymus, its chromosome includes only a subset of V, J and D units. That gives body the potential to create huge repertoire of different T-cells or B-cells to fight against various invasions. You can learn about the all genetic steps of above process here.
The next obvious question is how many different types of T-cells are present in human body. A mathematical limit can be computed by performing permutation/combination from the numbers of different Vs, Js and Ds, but that limit is too high. Our body does not allow many of those combinations to survive, because they are likely to attack our own organs. The adapters in some T-cells are such that they can recognize parts of our own body as foreign objects and destroy them. You do not want those deadly killers floating in the blood. Also note that the set of friendly T-cells vary from person to person. T-cells that are friendly to my body may not like organs transplanted from someone else’s body into mine.
It is estimated that typical human body carries something of the order of 10^ 6 different ‘good’ T-cells, but getting a proper count was impossible until high-throughput sequencing came along. The Fred Hutch team, led by Harlan Robins, used deep sequencing and multiplex PCR to sequence the V(D)J regions of almost all T-cells in a blood sample. Quoting from their paper -
The nucleotide sequence encoding the TCR? chain is determined by somatic rearrangement of V, D, and J segments along with a set of non-templated insertions and deletions at the junctions between the V and D, and the D and J segments. We have designed a pool of primers to all V and J pairs such that they specifically amplify the complete VDJ junction region, known as the CDR3 region, as well as a portion of the J and V segments that is sufficient for identification.
Finding T-cell diversity in different people (or in the same person at different times) is not only an intellectual question, but has immense practical use. Let us give you one example. Leukemia was considered a universally fatal disease 30 years back, but human society made great progress to cure it in at least some cases.
Leukemia is cancer of white blood cells, i.e. B-cells and T-cells. Quoting from wiki -
Acute leukemia is characterized by a rapid increase in the number of immature blood cells. Crowding due to such cells makes the bone marrow unable to produce healthy blood cells. Immediate treatment is required in acute leukemia due to the rapid progression and accumulation of the malignant cells, which then spill over into the bloodstream and spread to other organs of the body. Acute forms of leukemia are the most common forms of leukemia in children.
Relapse of the disease is a huge problem in leukemia treatment. Even after all cancerous cells are removed from blood, a very small number of bad ones survive. Those bad ones grow over time and bring back cancer in a person thought to be cured. The name of this problem is called minimum residue disease (MRD).
You can imagine that finding those small number of surviving cancerous cells is extremely difficult. It is not possible to identify them by visual inspection through microscope, because their numbers are close to one in a million. Their detection requires an extremely sensitive approach.
Deep sequencing happens to be the right approach for MRD. In a paper published in Science Translational Medicine, Dr. Robins and his team reported -
Here, we applied HTS to the diagnosis of T-lineage acute lymphoblastic leukemia/lymphoma. …..The variable regions of TCRB and TCRG were sequenced using an Illumina HiSeq platform after performance of multiplexed polymerase chain reaction, which targeted all potential V-J rearrangement combinations…..We found that TCRB and TCRG HTS not only identified clonality at diagnosis in most cases (31 of 43 for TCRB and 27 of 43 for TCRG) but also detected subsequent MRD. As expected, HTS of TCRB and TCRG identified MRD that was not detected by flow cytometry in a subset of cases (25 of 35 HTS compared with 13 of 35, respectively), which highlights the potential of this technology to define lower detection thresholds for MRD that could affect clinical treatment decisions.
For those who love challenges, the best place to learn about the above approach is these two patents - METHOD OF MEASURING ADAPTIVE IMMUNITY (2010) and METHOD OF MEASURING ADAPTIVE IMMUNITY (2012). Remaining souls should satisfy themselves with the following papers and two linked earlier -
Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells,
Deep sequencing of the human TCR? and TCR? repertoires suggests that TCR? rearranges after ?? and ?? T cell commitment, also pdf here.
-——————————————
Few other relevant links -
An ‘early’ paper from 1980. Yes, the first author is named Early, no kidding - An immunoglobulin heavy chain variable region gene is generated from three segments of DNA: VH, D and JH.
An old paper from PNAS 1993 that helps you track early progress in the field - The sizes of CDR3 hypervariable regions of the murine T-cells
An earlier paper on detection of MRD - Detection of Minimal Residual Disease in Acute Leukemia by Flow Cytometry
Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities
On spectratyping, which was the pre-NGS approach to study CDR3 diversity -
One of the first papers to use HTS to find antibody diversity High-Throughput Sequencing of the Zebrafish Antibody Repertoire
[The papers citing the above one are worth reading]
A 2011 paper that uses gene expression to find new markers for MRD detection
- New markers for minimal residual disease detection in acute lymphoblastic leukemia
A recent review on MRD diagnosis - Pretransplant MRD: the light is yellow, not red -Alan S. Wayne and Jerald P. Radich
-——————-
In the following post, we will cover the bioinformatics aspects of T-cell diversity profiling.