
Can Trinity be Used for Genome Assembly?
For new readers, easiest way to follow us is through our twitter feed. The feed is updated, whenever we post a commentary here.
A reader asked by private email whether Trinity can be used for genome assembly. With his permission, we are posting his question (edited to remove personal details) and our response here.
We should mention that his ‘newbie’ description caught us a bit off-guard at first. Based on his follow up question, it appeared that he used well-reasoned argument to try the mentioned approach. Few months back, we used the same rationale to run Trinity on one of our genomic libraries.
Question:
I am a newbie in transcriptome and genome assembly and
analysis. While browsing regarding the genome assembly i found your Homologous blog with many interesting topics and clear explanations.
I have a query regarding the usage of trinity and please spare me if i am not able to put it clearly.
I have genome data from Illumina platform of around ~8 Million PE reads and of 125bp length. I am supposed to assemble the reads for the further analysis. Since, genome and transcriptome assembly are not same, should not i use the RNA-seq assembler Trinity for making the DNA transcripts and remove the so called “alternative isoforms” from the Trinity.fasta and make use of contig file?
Would be thankful to your answer.
Our initial Response:
Here is the difference between genomic library and transcriptomic library. A Genomic library encounters reads extracted uniformly from the genome. So, when some reads (or parts thereof) appear in a genomic library with frequency higher than average, they are likely from repetitive regions of the genome.
Transcriptomes do not encounter much of repetitive regions. However, different genes have different expressions in a tissue, and so transcriptome libraries have reads coming in variable frequency because of data type.
Because of above differences, transcriptome assemblers tuned to handle transcriptomic reads may not do well with genomic data. Therefore, I think you should start with a genome assembler (Velvet, SOAPdenovo) etc. If you experiment with Trinity on genomic data, please make sure you understand the algorithm well to find/interpret any potential mis-assembly.
His follow up comment:
Thank you for the reply. But i would still like to know what is the
“tuning” done in terms of handling the transcriptomic reads? My point is,
The assembler does not distinguish the reads, whether they are C-DNA or DNA
and the point i am getting confused is, as long as the assembler is using
KMER concept to make the contig out of reads, why bother whether it should
be used for a particular type of OMIC assembly?
At that point, we decided to go to the drawing board and chalk up an example.
Here is a neighborhood from a very simplified genome. It contains two unique regions flanking one repetitive region appearing exactly twice in the genome. We note that real repeats are never so simple.
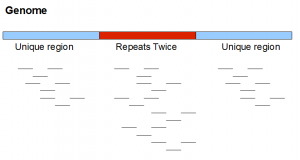
If the above genome is sequenced with short reads, short reads from the repetitive region will have twice as much frequency as in the unique region. As we mentioned, in real data repetitive regions are not always identical. So, short reads themselves may not show exactly twice the frequency. Only at a k-mer level, you may be able to see the second peak at frequency twice of the primary peak.
Here is a transcriptome experiment with two genes equally expressed at low level and a third gene expressed at level twice as that of other two genes. We also show the tentative RNAseq reads.
![]()
Do you notice some similarities between picture 1 and picture 2? Differential expression to a transcriptome assembler is conceptually similar to repeats to a genome assembler.
Anyway, getting back to our original question, all transcriptome assemblers first run a ‘genome assembly’ step to identify simple and contiguous segments. For Velvet+Oases pipeline, that step is done by Velvet genome assembler. For Trinity, the same step is run by ‘Inchworm’.
Long story short, if you use Trinity on genomic dataset and take the Inchworm output, that should be useful for genome assembly just like Velvet contigs. Trinity is cocktail of three independent programs - Inchworm, Chrysalis, and Butterfly. Inchworm follows a greedy algorithm to find contiguous segments in the data. Only in Chrysalis and Butterfly stages, Trinity refines the Inchworm output to define alternatively spliced genes as Velvet does with Oases.
However, I suspect Velvet will do better job than Inchworm. I need to recheck the algorithm but I believe Inchworm will not give you connections derived from paired end information. Those are used at a later stage of Trinity. From that viewpoint, Velvet may do a better job than Inchworm. If you have longer mate pairs, Trinity will definitely not take that into account like Velvet.
That is our simplest understanding of how the above programs work, and we are going to stick to it :)