
The World of Mapping Programs
Readers should not miss a highly informative website at EBI comparing all sequence alignment programs based on features, speed, memory requirement and citations, and places them on a ‘timeline chart. For convenience of readers, we present their timeline chart below, but here is the original link.
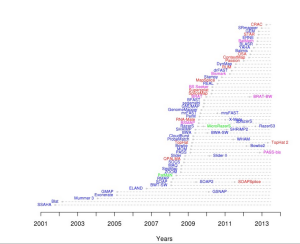
BLASR paper is at the bottom their citation table due to having no citation (too new), but we like it most for presenting a different perspective on the same search algorithms. Shown below is Fig. 1 from BLASR paper. It arranges the same mapping programs in a tree format created based on similarity of algorithms.

Figure 1 An illustration of relationships between alignment methods. The applications / corresponding computational restrictions shown are (green) short pairwise alignment / detailed edit model; (yellow) database search / divergent homology detection; (red) whole genome alignment / alignment of long sequences with structural rearrangements; and (blue) short read mapping/rapid alignment of massive numbers of short sequences. Although solely illustrative, methods with more similar data structures or algorithmic approaches are on closer branches. The BLASR method combines data structures from short read alignment with optimization methods from whole genome alignment.
If someone writes a book on sequence alignment, the above picture should be on its first page, because it describes the evolution of various algorithms in the most compact format.