Steven Salzberg at #bog13: Assembling 22Gb Conifer Genome

Steven Salzberg’s group at John’s Hopkins university is working on assembling the genome of a tree that is as tall as his publication list. He was kind enough to share the slides of his talk at the Biology of Genome 2013 conference taking place in Cold Spring Harbor Lab.
Few highlights of the talk based on his slides, private email exchanges and incredibly informative twitter updates from @leonidkruglyak, @ewanbirney and other #bog13 attendees:
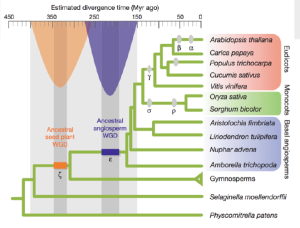
Gymnosperms are way out in the phylogenetic tree of plants, and the other two out-branches in the following figure are fern and moss. JGI already sequenced fern and moss genomes. Gymnosperm genome will help biologist understand genetic diversity of higher plants.
-
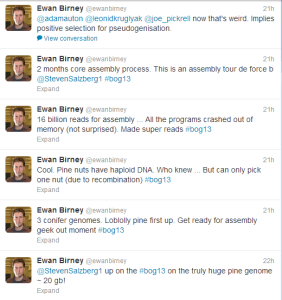
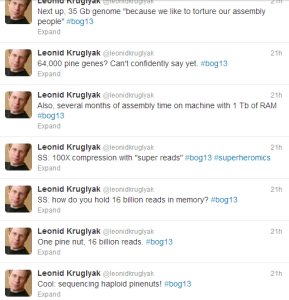
Here is (or was until NGS and SS arrived on scene) the problem = genome size of Loblolly pine is 22 Gigabases and that is not the largest of three pine genomes Salzberg group assembling. Another 35 Gb conifer monster is waiting at their door.
-
Most tissues in pine seeds are haploid megagametophyte.
In heterosporous plants (ferns, lycophytes, angiosperms and gymnosperms) the egg producing gametophyte is known as a megagametophyte and the sperm producing gametophyte as a microgametophyte. In the seed plants, the immature microgametophyte is called pollen. Seed plant microgametophytes consists of between two and five cells when the pollen grains exit the sporangium. The megagametophyte develops within the megaspore of extant seedless vascular plants and within the megasporangium in a cone or flower in seed plants. In seed plants, the typical microgametophyte (pollen grain) travels to the vicinity of the egg cell (carried by a physical or animal vector), and produces two sperm by mitosis.
-
Sequencing is being done by combining whole genome shotgun (WGS) and pooled cosmid clone strategies. So far the WGS part is complete.
-
Next thing you know, 16 billion Illumina paired reads (2x100 bp) arrived to crash his 1TB RAM server. Salzberg’s team decided to ‘compress’ the reads into 150 million super-reads.
-
Overall assembly took 10 days for error correction, 11 days for computing super-reads, 60+ days for contig and scaffold construction and 8 days for gap filling !!
-
The assembly was done using Celera assembler. For those who are wondering what SOAPdenovo would do, they presented a poster at #bog13. Hopefully we will get a copy to post it here soon.
-
The genome and RNAseq data gave them 64,000 protein coding genes. How real are them? A lot need to be sorted out on the biology front, which is not a surprise in a project of this scale.
-
What do bioinformaticians do, when their programs take months to run? Apparently Steven Salzberg spent time acting in French movies:
We had few more questions and Dr. Salzberg kindly helped us out.
Question: With this big genome, I would assume the sequencing is highly non- uniform over the genome. So, essentially you have many problems faced by meta- genome assembly group. Do you need to factor them in?
SS: sequencing seems to be quite uniform which is good. Size does not correlate with non-uniformity so this isn’t particularly surprising.
Question: Where are the current assembly stats?
SS: I gave the main ones in the talk - contig N50 is 8.2 kilobases, scaffold N50 is 31-32 Kb, and assembled contigs sum to 20.1 Gbp.
Question: 22 GB would mean hell of an amount of repeats. Do you estimate how much of the genome you will be able to assemble after excluding them?
SS: Everyone assumes lots of repeats. Of course they are there, but they are not so identical to one another that they cause a major problem. Some problems, yes, but we are able to resolve most repeats and separate them.
What biology are you guys looking at in addition to being able to place gymnosperms at the right place? Is the genome necessary, or would the time/money have been better invested sequencing the transcriptomes of many pines, ferns and mosses? I do not meant to undermine the incredible job of assembly you guys are doing, but rather trying to see the bigger biological context.
**SS: lease check out PineRefSeq for lots and lots of info about pines and their importance in biology, evolution, and our economy: http://pinegenome.org/pinerefseq/.
**
Edit. Dr. Salzberg requested us to include the following passage. For more information, please check this commentary.
One important point overlooked in the commentary was that University of Maryland (WMD) did most of the work on the whole-genome assembly with MaSuRCA, and the acronym itself stands for Maryland Super-Reads with Celera Assembler. The leaders of the UMD effort are Aleksey Zimin and Jim Yorke.
Please click on the image to see the slides:

Here are some relevant tweets and an earlier PLOS One paper mentioned by Surya Saha -