Genome Wide Association Studies (#GWAS) - Are They Replicable?
Criticism of genome wide association studies got two Dans into twitter brawl. Such exchanges are always good opportunities to learn.
We have our own biases on the issue, but keeping that aside, let us see what the others are saying.
The paper under consideration is here -
The authors set to reproduce the results of a commonly-cited GWAS study on obesity.
Introduction:
Obesity is a major risk factor for many non-communicable diseases including type 2 diabetes, cardiovascular disease, and certain cancers [1]. Genetic predisposition and lifestyle factors are known to increase obesity susceptibility, and the technological breakthroughs that came with genome-wide association studies (GWAS) have led to the successful identification of a large number of obesogenic loci [2][6]. Recent studies suggest that physical activity may modify genetic susceptibility to obesity, with the genetic burden being higher in physically inactive compared with active persons [7][9]. The most extensively studied example of a gene physical activity interaction in obesity is for the FTO locus [7], [10], which was recently replicated in a meta-analysis comprising 240,000 persons [11]. Elsewhere, Li et al reported that physical activity offsets the aggregated genetic risk of 12 obesogenic loci [12].
In the current study, we aimed to replicate the findings of Li et al [12] in a sample collection of 111,421 individuals of European ancestry. We also undertook detailed analyses focused on the role of within- and between-study factors to establish how the design of gene environment interaction meta- analyses impacts the power to detect interactions.
What did they find?
In summary, our meta-analysis of 111,421 samples from 11 cohorts of European ancestry yielded results that support those of Li et al [12]. However, these effects appear evident only when the cohorts from North America (n = 39,810) are included in this meta-analyses. We also demonstrate using simulated data that combining many small cohorts that vary in their classification of physical activity and other factors is a relatively inefficient approach to studying interactions; hence, future studies of gene lifestyle interactions might prove most effective if focused on a small collection of large cohorts within which standardized and valid lifestyle assessment methods are available.
That was easy. The authors claimed that they reproduced the results of Li et al. So, apparently Dan Graur should complain only after taking GWAS101 class from from Dan MacArthur.
-———————————————————
Well, not so fast. The following twitter discussion shows the real problem.
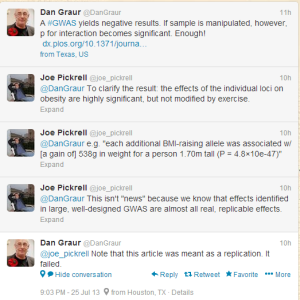
See, to make sure that a scientific result is valid, you have to give a group of unbiased researchers or qualified technicians a paper and ask them to repeat the reported steps in the study. If they follow the steps, but cannot reproduce the result, then the original study is not reproducible. However, if you, the author of the original study, are allowed to make suggestions during the replicate experiment about few steps here and there, the whole scientific process gets broken and bias is introduced.
To understand the difference between previous and current study, we need to take a look at the abstract of the previous study.
Methods and Findings
We genotyped 12 SNPs in obesity-susceptibility loci in a population-based sample of 20,430 individuals (aged 3979 y) from the European Prospective Investigation of Cancer (EPIC)-Norfolk cohort with an average follow-up period of 3.6 y. A genetic predisposition score was calculated for each individual by adding the body mass index (BMI)-increasing alleles across the 12 SNPs. Physical activity was assessed using a self-administered questionnaire. Linear and logistic regression models were used to examine main effects of the genetic predisposition score and its interaction with physical activity on BMI/obesity risk and BMI change over time, assuming an additive effect for each additional BMI-increasing allele carried. Each additional BMI-increasing allele was associated with 0.154 (standard error [SE] 0.012) kg/m2 (p = 6.7310?37) increase in BMI (equivalent to 445 g in body weight for a person 1.70 m tall). This association was significantly (pinteraction = 0.005) more pronounced in inactive people (0.205 [SE 0.024] kg/m2 [p = 3.6210?18; 592 g in weight]) than in active people (0.131 [SE 0.014] kg/m2 [p = 7.9710?21; 379 g in weight]). Similarly, each additional BMI-increasing allele increased the risk of obesity 1.116-fold (95% confidence interval [CI] 1.0931.139, p = 3.3710?26) in the whole population, but significantly (pinteraction = 0.015) more in inactive individuals (odds ratio [OR] = 1.158 [95% CI 1.1181.199; p = 1.9310?16]) than in active individuals (OR = 1.095 (95% CI 1.0681.123; p = 1.1510?12]). Consistent with the cross-sectional observations, physical activity modified the association between the genetic predisposition score and change in BMI during follow-up (pinteraction = 0.028).
Conclusions
Our study shows that living a physically active lifestyle is associated with a 40% reduction in the genetic predisposition to common obesity, as estimated by the number of risk alleles carried for any of the 12 recently GWAS-identified loci.
Do you see the problem? The old study was about Europeans, but the new study could not reproduce it until ~40,000 Americans were added in.
However, a statistically significant interaction effect was only apparent in North American cohorts (n = 39,810, Pinteraction = 0.014 vs. n = 71,611, Pinteraction = 0.275 for Europeans).
You may say that Americans are of European origin as well, and therefore there is nothing wrong to add them. That is exactly what the new study did. The broadened the scope to ‘Individuals of European Ancestry’ to claim that they reproduced the result. However, it is actually a new result, and has to be validated by another replication study.
New scientific results are always exciting, but here is the problem in the bigger context. See how much the society spent to go nowhere, because the above study needs to be replicated again by someone else before we can trust its results. The authors changed the goal-posts to call their replication a valid ‘goal’.
Funding: Some of the work leading to this publication benefited from support from the Innovative Medicines Initiative Joint Undertaking under grant agreement n115317 (DIRECT), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/20072013) and EFPIA companies’ in kind contribution. The current study was funded by Novo Nordisk, the Swedish Research Council, Phlssons Foundation, the Swedish Heart-Lung Foundation, and the Skne Regional Health Authority (all to PWF). The Fenland Study is funded by the Wellcome Trust and the Medical Research Council (MC_U106179471). The present part of the HEALTH2006 study was funded by The Lundbeck Foundation Centre for Applied Medical Genomics in Personalised Disease Prediction, Prevention and Care (LuCamp, www.lucamp.org). The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk). The present part of the Inter99 study was funded by The Lundbeck Foundation Centre for Applied Medical Genomics in Personalised Disease Prediction, Prevention and Care (LuCamp, www.lucamp.org). The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk). InterAct study was supported by funding from the European Union (Integrated Project LSHM-CT-2006-037197 in the Framework Programme 6 of the European Community) and the Medical Research Council, UK. The MDC study was supported by research grants from Swedish Research Council, the Swedish Heart-Lung Foundation, Strategic Research grant to EXODIAB, Linneus grant to Lund University Diabetes Centre (LUDC), The Albert Phlsson Foundation, The Novo Nordisk Foundation, The Swedish Diabetes Foundation and an equipment grant from The Knut and Alice Wallenberg Foundation. METSIM study: This work has been supported by the Academy of Finland, the Finnish Diabetes Research Foundation, the Finnish Cardiovascular Research Foundation, EVO grant from the Kuopio University Hospital (5263), NIH grants DK093757, DK072193 and DK062370. The NHS study was supported by grants DK091718, HL071981, HL073168, CA87969, CA49449, CA055075, HL34594, HL088521, U01HG004399, DK080140, 5P30DK46200, U54CA155626, DK58845, U01HG004728-02, EY015473, DK70756, CA134958, and DK46200 from the National Institutes of Health, with additional support for genotyping from Merck Research Laboratories, North Wales, PA. The Swedish Twin Registry is supported by the Ministry for Higher Education, the Swedish Research Council, and GenomEUtwin; the US National Institutes of Health; and the Swedish Foundation for Strategic Research. EI and AG were supported by the Swedish Heart-Lung Foundation (grant no. 20120197) and Swedish Research Council (VR; grant no. 2012-1397) when working on this paper. The WGHS is supported by HL043851 and HL080467 from the National Heart, Lung, and Blood Institute and CA047988 from the National Cancer Institute, the Donald W. Reynolds Foundation and the Fondation Leducq, with collaborative scientific support and funding for genotyping provided by Amgen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
-——————————————-
Which of the two Dans is correct in the bigger context of #GWAS (not specific to the mentioned paper)? Both of them actually, as argued by Mary-Claire King. Dan MacArthur likely assumes that genome-wide association studies are being criticized, because they have methodological flaws. That is not correct, as Dr. King pointed out in the text presented below.
Genetic Heterogeneity in Human Disease
The general failure to confirm common risk variants is not due to a failure to carry out GWAS properly. The problem is underlying biology, not the operationalization of study design. The common diseasecommon variant model has been the primary focus of human genomics over the last decade. Numerous international collaborative efforts representing hundreds of important human diseases and traits have been carried out with large well-characterized cohorts of cases and controls. If common alleles influenced common diseases, many would have been found by now. The issue is not how to develop still larger studies, or how to parse the data still further, but rather whether the common diseasecommon variant hypothesis has now been tested and found not to apply to most complex human diseases.
In the bigger context of curing complex diseases, finding the associations is a step toward finding the genetic mechanisms behind the disease and then that will be followed by targeted drug design. It is just that, compared to their costs, the #GWAS studies are not producing enough biological results or insights. In Mary-Claire King’s words, “it is now clear that common risk variants fail to explain the vast majority of genetic heritability for any human disease”.
Mary-Claire King does not have to be always right. To balance our potential bias in the above description, we will allow Dan MacArthur to have the final word -