
On FPGA Acceleration of Bioinformatics Programs
We have been working on FPGA acceleration of one or two bioinformatics programs and, during the process, thinking of ways to do the same for many other programs. The problems are numerous and we would like to share with you, what we learned so far.
If you read the literature, you will find several papers describing how to accelerate this or that algorithm in FPGA. Those papers are often written by electrical engineers and get published in some VLSI journal, but rarely reach typical biology labs.
Why not? It is because the biologists/bioinformaticians like to use certain ‘standard’ pipelines, and will complain, if they do not see the same exact program producing the same exact output being accelerated. It is not entirely their fault. Often the larger pipelines are written and tested with one program and cannot be made to run easily with a different one. Why doesn’t everyone use BWA-Tophat-Cufflink? Why doesn’t RSEM get run with SOAP2 and and GATK with Bowtie? For a good real life example, check Rayan’s post in Biostar, where he used SSPACE configured with Bowtie and BESST with BWA-MEM. Theoretically, it should be straightforward to interchange two aligners or two scaffolders in his analysis. It is also theoretically possible to make a pig fly, because all its atoms moving upward at the same time has a finite probability in quantum mechanics.
Getting back to the FPGA-acceleration dilemma, the solution is to accelerate standard or commonly used program. That is easy to say, but quite difficult to execute at scale. Most commonly used programs are written for CPU architecture and simpler ones can be described in the following manner.
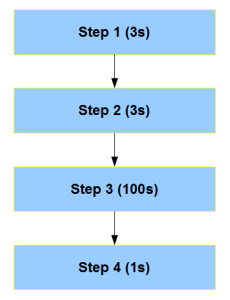
Let us say, you make Step3 100 times faster in FPGA. Then all the equations change, because Step3 is now the fastest piece in the whole chain and not rate-limiting.
Moreover, above figure is one of the simpler situations. Most complex bioinformatics codes have multiple critical blocks and loops within each block. Therefore, accelerating one entire block or multiple synchronized blocks is not easy. The best way to proceed is to build the design bottom up from the FPGA through multiple interacting threads, but that is not how the CPU code is written. On the other hand, the restriction that the program produces the same output as before means one needs to view the standard program top down as well. Combining the ‘bottom up’ and ‘top down’ criteria will essentially make you rewrite the entire code - not a happy prospect in many situations. No matter what, accelerating a program in FPGA means you need to understand the algorithm in minute detail and not just peripherally (at block diagram level).
We will share further insights as we continue to work on this problem. BGI is possibly one of the few organizations looking into hardware (GPU) acceleration of commonly used NGS bioinformatics algorithms, and we have been reading their papers and reports with great interest.