
Kmer Counting - a 2014 Recap
Kmer counting is the easiest problem in bioinformatics. Any novice with a few minutes of training in PERL or python can write a k-mer counting program. It is so simple that when we were learning Hadoop, we chose k-mer counting as the warm up exercise.
Kmer counting is also the hardest problem in bioinformatics, especially if someone has to scale for billions of reads. We wrote about a number of elegant k-mer counting codes in 2012 -
Efficient Methods for Counting K-mers
Since then, the following papers or releases came out.
DSK: K-mer Counting with Very Low Memory Usage
Efficient Online k-mer Counting using a Probabilistic Data Structure
Compressed Gk Arrays for K-mer Counting [Please see Rayan’s warning in the post]
Now that it is 2014, time to review the field again.
-———————————————————–
Why is k-mer counting difficult? It is because the k-mer space is huge and its size increases exponentially with k (=4^k). Given that the space is also sparsely occupied, one would immediately think about hashing as a way to solve the problem.
That strategy poses one difficulty. In case of k-mer counting, people do not only want the counts, but also the original k-mers. Hash functions are usually not reversible.
How do various programs solve this?
Jellyfish
Jellyfish is a hash-based counting method that stores both the count and the k-mer in the hash location. In case of collision, it finds a new location for k-mer and count. If the table is too full, Jellyfish dumps counts in disk and merges later. The biggest contribution of Jellyfish is possibly its use of ‘compare-and-swap’ memory update. That way multiple threads can count k-mers in parallel without need to lock memory update step.
BFCounter
BFCounter uses the idea that singleton k-mers are the most abundant in next- gen libraries of genomic origin, and gets rid of them through a Bloom filter.
CM-Sketch (Titus Brown)
CM Sketch is appropriate when the exact k-mer count is not needed and some degree of approximation is ok. It uses multiple hash tables to store the count of each k-mer, but not the k-mer. Recovery of count of each k-mer is done by probing the corresponding locations of all hash tables and taking the minimum of the counts.
DSK
DSK stores all k-mers in multiple files in the disk and then counts each file one by one using a hashing strategy. Since storing all k-mers of a large library requires humongous amount of space, DSK often needs multiple passes through the read library.
Edit. Rayan mentioned KMC in comment section. The method of KMC appears to be similar to DSK.
Very recently, a disk-based k-mer counting algorithm, named DSK, was presented [9]. On the high level, DSK is similar to the solution presented in this paper, but both algorithms were designed independently at about the same time. In DSK, the (multi)set of k-mers from the input reads is partitioned and partitions are sent to disk. Then, each partition is loaded and processed separately in the main memory, using a hash table. The tool provides strict control of the allocated memory and disk space (lower memory usage results in increased processing time due to more iterations and thus increased I/O), which for example allows to process human genome data in 4 GB of RAM only, in reasonable time.
Also check scTurtle paper in the same context.
MSPKmerCounter
Unpublished MSPkmercounter is similar to DSK except that it uses the idea of minimizers in splitting the reads into ‘super-kmers’. That saves space and memory for subsequent processing.
-———————————————————–
Edit.
We are adding the following ones based on comments from readers. We have not had a chance to think deeply about their algorithms.
KmerStream
This algorithm from Pall Melsted just came out in bioRxiv.
Results: We present KmerStream, a streaming algorithm for computing statistics for high throughput sequencing data based on the frequency of k-mers. The algorithm runs in time linear in the size of the input and the space requirement are logarithmic in the size of the input.
Visualizing the Repeat Structure of Genomic Sequences
Nava E. Whiteford added a paper of him. It uses suf?x array and LCP array.
KAnalyze: A Fast Versatile Pipelined K-mer Toolkit
Peter Audano pointed out in the comment section about one of his papers.
Motivation: Converting nucleotide sequences into short overlapping fragments of uniform length, k-mers, is a common step in many bioinformatics applications. While existing software packages count k-mers, few are optimized for speed, offer an API (Application Programming Interface), a graphical interface, or contain features that make it extensible and maintainable. We designed KAnalyze to compete with the fastest k-mer counters, to produce reliable output, and to support future development efforts through well architected, documented, and testable code. Currently, KAnalyze can output k-mer counts in a sorted tab-delimited file or stream k-mers as they are read. KAnalyze can process large data sets with 2GB of memory. This project is implemented in Java 7, and the CLI (Command Line Interface) is designed to integrate into pipelines written in any language.
Results: As a k-mer counter, KAnalyze outperforms Jellyfish (Marais and Kingsford, 2011), DSK (Rizk et al., 2013), and a pipeline built on a Perl and Linux utilities. Through extensive unit and system testing, we have verified that KAnalyze produces the correct k-mer counts over multiple data sets and k-mer sizes.
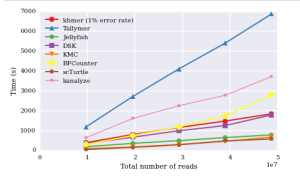
Titus Brown claims it is slower than others, and posted this ipython notebook.
One of his comparative chart is shown below. The rest are available from the posted ipython notebook link.
-—————————————————
Did we miss anything? Oh well, we missed mentioning that k-mer counting and BWT construction are equivalent processes, as explained here.
BWT Construction and K-mer Counting
Does that mean someone will be able to apply minimizers to develop an efficient BWT construction algorithm?