
Latest from Tony-Cox - BEETL-fastq
Compressing and storing of Illumina libraries is a major problem. A number of groups are working on BWT/FM-index-based solutions, so that the compressed file can be searched easily. We already reported about Heng Li’s “Fast construction of FM-index for long sequence reads” and BGI’s GPU-based solution, but Tony Cox, the earliest contributor to this area, is not sitting on his hand.
BEETL-fastq: a Searchable Compressed Archive for DNA Reads
Motivation: FASTQ is a standard file format for DNA sequencing data which stores both nucleotides and quality scores. A typical sequencing study can easily generate hundreds of gigabytes of FASTQ files, while public archives such as ENA and NCBI and large international collaborations such as the Cancer Genome Atlas can accumulate many terabytes of data in this format. Text compression tools such as gzip are often employed to reduce the storage burden, but have the disadvantage that the data must be decompressed before it can be used. Here we present BEETL-fastq, a tool that not only compresses FASTQ-formatted DNA reads more compactly than gzip, but also permits rapid search for k-mer queries within the archived sequences. Importantly, the full FASTQ record of each matching read or read pair is returned, allowing the search results to be piped directly to any of the many standard tools that accept FASTQ data as input.
Results: We show that 6.6 terabytes of human reads in FASTQ format can be transformed into 1.7 terabytes of indexed files, from where we can search for 1, 10, 100, 1000, a million of 30-mers in respectively 3, 8, 14, 45 and 567 seconds plus 20 ms per output read. Useful applications of the search capability are highlighted, including the genotyping of structural variant breakpoints and in silico pulldown experiments in which only the reads that cover region of interest are selectively extracted for the purposes of variant calling or visualization.
Availability:
BEETL-fastq is part of the BEETL library, available as a github
repository at git@github.com:BEETL/BEETL.git.
Contact: acox@illumina.com
Most importantly Cox now has a detailed flow diagram for operations, when his module is implemented in cloud.
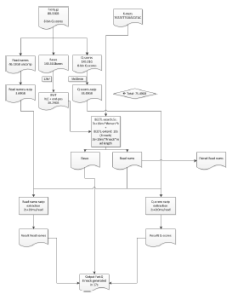
Illumina also applied for a patent in this algorithm, which possibly made BGI decide to patent genome assembly and PacBio to patent BLASR. Then Illumina lawyers made their final (and most laughable) move by deciding to patent all online bioinformatics programs. We will check with Dr. Cox about the status of the patent application of BEETL.
Keith Robison also pointed out -
We had a discussion on this issue with Tony Cox one year back. Readers may look at the comment section of this thread.