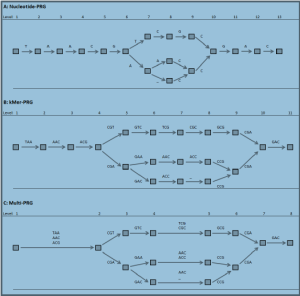
Kmers Rock - Improved Genome Inference in the MHC using a Population Reference
This new paper appeared in biorxiv (h/t: Jared Simpson). As always, the supplementary section has the most meat. The authors love kmers so much that they came up with new English words (kMerification, kMerify) and new way to write it (kMer).
In humans and many other species, while much is known about the extent and structure of genetic variation, such information is typically not used in assembling novel genomes. Rather, a single reference is used against which to map reads, which can lead to poor characterisation of regions of high sequence or structural diversity. Here, we introduce a population reference graph, which combines multiple reference sequences as well as catalogues of SNPs and short indels. The genomes of novel samples are reconstructed as paths through the graph using an efficient hidden Markov Model, allowing for recombination between different haplotypes and variants. By applying the method to the 4.5Mb extended MHC region on chromosome 6, combining eight assembled haplotypes, sequences of known classical HLA alleles and 87,640 SNP variants from the 1000 Genomes Project, we demonstrate, using simulations, SNP genotyping, short-read and long-read data, how the method improves the accuracy of genome inference. Moreover, the analysis reveals regions where the current set of reference sequences is substantially incomplete, particularly within the Class II region, indicating the need for continued development of reference-quality genome sequences.