
Bioinformatics at the Core Facilities
Jeffrey Chang, the co-director of the Bioinformatics Service Center at University of Texas Health Science Center, wrote an informative comment in Nature and explained the challenges faced by core facilities offering bioinformatics services.
To give greater support to researchers, our centre set out to develop a series of standardized services. We documented the projects that we took on over 18 months. Forty-six of them required 151 data-analysis tasks. No project was identical, and we were surprised at how common one-off requests were (see ‘Routinely unique’). There were a few routine procedures that many people wanted, such as finding genes expressed in a disease. But 79% of techniques applied to fewer than 20% of the projects. In other words, most researchers came to the bioinformatics core seeking customized analysis, not a standardized package.
Another issue is that projects usually become more complex as they go along. Often, an analysis addresses only part of a question and requires follow-up work. For example, when we unexpectedly found a weak correlation between a protein receptor and a signalling response, getting a more-robust answer required refining our analysis to accommodate distinct signalling pathways orchestrated by the protein. An expansion typically doubled the time spent on a project; in one case it increased the time fivefold.
Believe it or not, we anticipated the same problems three years back, when we wrote -
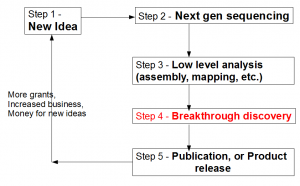
Let us view the entire loop (see figure above) from the viewpoint of a biologist. A biologist gets a novel idea (Step 1), and based on that, he contracts with a sequencing facility to sequence his samples. Sequencing center turns his sample into large library of A, T, G, Cs (Step 2). That sentence summarizes many complex steps, among which, sample preparation itself, can be quite challenging, especially for unusual experiments. Moreover, the efforts of sequencing facility are not straightforward by any means, because it has to keep abreast of changes in technologies, optimize protocols to get the highest throughput, get trained on latest protocols for unusual sequencing requests, upgrade software, and so on.
Step 3 requires converting the raw data into something more tractable. Typically, it involves mapping of reads on to a reference genome (if reference genome exists), or assembling of reads into larger units (contigs). This step is technologically very challenging due to the size of NGS data sets. The bioinformatician needs to filter reads for sequencing errors, try out various k-mer sizes, keep up with the latest analysis approaches, upgrade computers and RAMs for progressively larger libraries generated by new sequencing machines and so on.
What frustrates biologists the most is that even after three complex steps described above, they are no closer to the reward than months before. The next step breakthrough discovery is what ultimately makes their research visible and attracts funding for the efforts. For truly great discoveries, often the pipeline shown above is not linear. The scientists need to understand the results from step 3, go back to do more sample preparation, sequencing and assembly (iterations of steps 1-3). However, when multiple rounds of sequencing are done, analysis process does not remain straightforward as described in step 3. Integration of data generated from different rounds/kinds of sequencing and finding their cross-correlation becomes more difficult than assembling one round of data set.
As a simple example, let us say a research group did one round of RNAseq on five tissues and assembled genes using Velvet+Oases. Based on their analysis of the reads, they decide to run another round of RNAseq and assemble using Trinity. Should the next assembly merge all reads (old and new) together, and generate new sets of genes? How to deal with previous Oases genes and all expression-related downstream analysis done on the old set? Portability of data requires mapping of all old information on to new releases of genes and genomes. Every iteration adds layers of complexities, because often integration of old data necessitates redoing old calculations. One may say that the above difficulties can be overcome by proper planning, but think about challenges posed by publication of new and powerful algorithm that allows a more refined analysis of existing data. Or what if a group performs transcriptome experiment of large number of RNA samples and another group publishes the underlying genome in the meanwhile? It is nearly impossible to prepare for all eventualities in such a fast-moving field.
What is the solution? We believe bioinformatics analysis cannot be done effectively as ‘core service’. Instead the researchers themselves will have to get their hands dirty and take control of their own data. The biggest challenge we encountered in working with hundreds of scientists is that a bioinformatician needs to fully appreciate the scientific problem being solved to effectively help a scientist. That means he needs to read the relevant papers and get to understand the issues at similar level as the involved researchers. It is impossible for a member of core facility to survey tens or hundreds of areas of biology and talk at expert level with similar number of biologists.
Going forward, we expect the core facilities to provide the infrastructure to run big calculations (see Lex Nederbragt’s A hybrid model for a High- Performance Computing infrastructure for bioinformatics), or provide training on new software, but beyond that we do not expect them to be of much benefit in doing real research. In fact, focusing too much on centralized services will result in delay and reduction of quality.