Why Don't Transcription Factors Get Lost in Huge DNA Maze?
Mike White had more discussion in his blog on ‘the paper that should not be published anywhere’, and it is quite thought-provoking.
Imagine you have to go into a library with over 3 billion ‘randomly arranged’ books to find five or ten books of your interest. That task is extremely difficult if not impossible. Transcription factors in cell do something similar, when they find five, ten or fifty genes they like to regulate. We manage to find things in libraries, because they are nicely indexed. Transcription factors are known to bind to motifs, which are too short for indexing the 3 Gb genome. How do they ever manage to find the locations of genes they regulate?
Mike had two hypotheses.
A. Most of those 3 billion books are worn out magazines and the library management decided to keep them locked somewhere. Only 300 million books are accessible to you and you know that your books are there. The task is still humongous, but less difficult than looking through 3 billion books.
In Mike’s language,
Hypothesis A: Chromatin context is everything. In any given cell, most of the genome is inaccessible, wrapped up into large, compact regions of dense chromatin. This reduces the transcription factors search space, so they dont get lost.
But if context is the answer, how do the right parts of the genome get left exposed?
B. Transcription factors index the genome using longer motifs, but we do not know about it.
Hypothesis B: DNA grammar: short 8-base pair recognition sequences are not enough; true functional sites consist of rare, highly specific combinations of short recognition sites. The millions of spurious sites in the genome do not have the right DNA grammar, and they are not bound.
But if grammar is the answer, why do transcription factors seem to non- specifically bind all over the place?
‘The paper that should not be published anywhere’ did an experiment. Mike was thinking that A would be correct and the result came out to be of surprise to him.
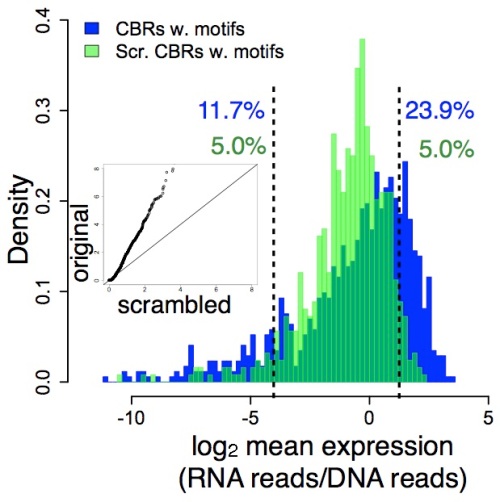
We tested large numbers of bound and bound DNAs in our massively parallel1 functional (i.e., reporter gene) assay. (We did this in the dissected whole retinas of baby mice.) The result came as a surprise to me, because I was betting on Hypothesis A. But it turns out that bound DNA differed from our random DNA distribution (showing function), while the unbound DNA largely resembled the random DNA distribution (showing non-function).
You can see this in these histograms from Fig. 1 of our paper. In the first panel, bound DNA distribution is in blue, the random DNA distribution is in green, and the level of gene expression is shown on the x-axis:
In the next panel, unbound DNA is in blue, and random DNA is in green notice that these two largely overlap (except in the left tail):
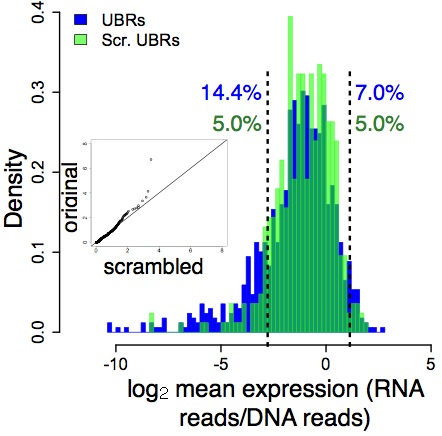
What this means is that the distinction between functional (bound) and non- functional (unbound) DNA is independent of context, at least to a large degree. The information that distinguishes function from non-function is therefore locally encoded in the short DNA regions (84 bases) that we tested.
Does that provide support for Mark Ptashne, who presented epigenetics and inheritance of acquired characteristics as faddish staff?
Somehow, the following ideas caught fire, even entering the standard textbooks: nucleosome modifications not only determine states of gene expression, but those modifications can be copied to maintain states of gene expression. The modifications were called epigenetic, implying that they convey self-perpetuating information as cells divide.
The problem with this characterization is that overwhelmingly, experiments have shown it to be false: histone modifications are not maintained as cells divide. These modifications turn over rather rapidly, and the nucleosomes themselves are too labile to carry information across cell divisions (2,,6). Moreover, there is no plausible molecular mechanism by which a gene that is on could be maintained on by such modifications (7).
Why then do daughter cells tend to mimic their mothers in their states of gene expression? There is no mystery: genes are activated (caused to be transcribed) by regulatory proteins, called transcription factors, which bind to DNA and work on nearby genes (7, 8). Exogenously added genes encoding regulatory proteins can cause somatic cells to change their identities, even forming pluripotent stem cells (9, 10), and the modern study of development entails, to a very large extent, the action of these proteins (11, 12).
Regulatory proteins in mother cells are, as a matter of course, distributed to daughter cells, where they bind DNA and turn on the same genes they activated in the mother cell (8, 13). Indeed, for a mother cell to give rise to two different daughters requires, in general, sequestration of one or more regulatory proteins (14), or the two daughter cells must receive from the environment different signals to which the regulatory proteins respond. In bacteria and in eukaryotes, the continuing presence of the activator is required to keep the gene on.
We will not know the answer until someone manages to define ‘epigenetics’ as precisely as other scientific terms, such as ‘mass’, ‘energy’ or ‘standard meter’.
Edit.
We checked with Mike While to make sure we are interpreting his results correctly. Here is the exchange.