
Debunking ENCODE's Latest Propaganda Piece Para by Para
ENCODE leaders published their latest propaganda piece in PNAS -
“Defining functional DNA elements in the human genome”
and Dan Graur has done fantastic work of tearing it apart.
Instead of rewriting his blog post, let us comment on random samples from here and there in the article.
-————————————————————–
Marketing Your Science, LLC
We never had honor of writing papers with co-authors from ‘scientific’ organizations like above. WTF - ‘science’ (i.e. an effort to find truth) needs marketing these days to get funded by government? Are those marketing efforts also paid by ENCODE/NHGRI?
That leads to the questions of how much of the PNAS paper is science and how much is ‘advertising’ (i.e. half truths and systematic efforts to hide the negatives)?
-————————————————————–
Proposed Future plan
In the last paragraph, ENCODE tells us what their next ‘big science’ scheme is going to be.
The data identify very large numbers of sequence elements of differing sizes and signal strengths. Emerging genome-editing methods (113, 114) should considerably increase the throughput and resolution with which these candidate elements can be evaluated by genetic criteria.
That is downright scary !! The ENCODE buffoons ‘proved’ 80% of human genome to be biochemically functional (whatever that means), which also implies that they will be allowed to waste the largest amount of money to do CRISPR-cas9 editing of nearly the entire human genome.
Speaking of CRISPR, we should also keep in mind that the discovery came from a French food company and not through NIH funds. If you read about the discovery in above link, you will realize that such innovative research projects are in fact at great disadvantage at NIH due to ENCODE-like wasteful and human-centric big science projects. Who cares about the fight between bacteria and phage, which has little ‘human connection’?
-————————————————————–
On Functional Elements
ENCODE clowns wrote -
Despite the pressing need to identify and characterize all functional elements in the human genome, it is important to recognize that there is no universal definition of what constitutes function, nor is there agreement on what sets the boundaries of an element.
Readers should note that the ‘pressing need’ claim came from ENCODE ‘leaders’ and nobody else. They were the ones, who invented this fictitious make-work project. Let us go through a bit of history. In 2003, our group at NASA (PI - Victor Stolc) and a Yale group involving two would-be ENCODE leaders published a tiling array scan of the entire human genome in Science.
Global identification of human transcribed sequences with genome tiling arrays
It was one of our most expensive projects at that time (~$100K of arrays). In fact, for much much less money, we made a number of other innovative studies in a range of model organisms. Cost of our very cool chlamy paper was only $4K of arrays and identified a set of potential regulators in human eye. Our yeast paper had a cost of around the same ($6K), sea urchin a bit more ($29K) and so on, and each experiment was a breakthrough compared to the status quo in those organisms. We often wondered whether the human effort was worth the money in comparison, given how much more we learned from experiments in other organisms. Little did we realize that Snyder/Gerstein would get to flip it to the government to waste $300 million dollars for almost no science !
ENCODE’s own website presents the following purpose for their project -
The National Human Genome Research Institute (NHGRI) launched a public research consortium named ENCODE, the Encyclopedia Of DNA Elements, in September 2003, to carry out a project to identify all functional elements in the human genome sequence.
That is from the first paragraph of overview in the website, which is from the first section of the entire website. After telling everyone for a decade that ENCODE’s goal was to find ‘functional elements’, now the buffoons inform us that it is impossible to define either ‘functional’ or ‘element’. That IS simply priceless !!!
Some of these differences stem from the fact that function in biochemical and genetic contexts is highly particular to cell type and condition.
Oh geez, did you just figure that out? And your solution was to take as many cell-types as possible and add up all effects to claim as much human genome to be as functional to write eulogy for junk DNA, right?
Moreover, each approach remains incomplete, requiring continued method development (both experimental and analytical) and increasingly large datasets (additional species, assays, cell types, variants, and phenotypes).
Well, one big thing that is complete is the number 100. Once you reach 100% of the genome, we hope it will not be necessary to look further for functional elements.
-————————————————————–
Low, Medium and High Evidence
A 2012 press release from Zhiping Weng, one of the ENCODE leaders, said -
Using data generated from 1,649 experimentswith prominent contributions from the labs of Job Dekker, PhD, professor of biochemistry & molecular pharmacology and molecular medicine, and Zhiping Weng, PhD, professor of biochemistry & molecular pharmacologythe group has identified biochemical functions for an astounding 80 percent of the human genome. These findings promise to fundamentally change our understanding of how the tens of thousands of genes and hundreds of thousands of gene regulatory elements, or switches, contained in the human genome interact in an overlapping regulatory network to determine human biology and disease.
Note the words - ‘identified biochemical functions for an astounding 80 percent of the human genome’, ‘fundamentally change our understanding’.
Here is the main figure of the new PNAS paper -
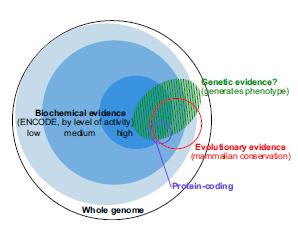
It suggests that the previously marketed astounding discovery was backed by low evidence data. Not only that, the figure caption of the new figure has the audacity to say -
This summary of our understanding in early 2014 will likely evolve substantially with more data….
So, we now learn that $300 million later, ENCODE did not manage to generate enough ‘data’ to back a point, which they previously claimed to be true in numerous press releases. It is as if NASA goes around telling everyone that that they heard voices from humans living in Urenus, and reveal on further prodding that the claim was all based on low-quality information.
Such a thing can only happen in the ENCODE world !
-————————————————————–
** GATA1 - Small Science versus Big Science **
The paper discusses one gene - GATA1 in quite a bit of detail. Why GATA1? A bit of googling tells us that it is the gene Ross Hardison, the corresponding author, spent years working on and understands well. That is a product of ‘small science’, which is supposedly discredited by ENCODE-type large-scale efforts.
In fact, resorting to a familiar gene disproves everything ENCODE stands for. If big science worked, ENCODE team could have easily pulled an arbitrary segment of genome they knew nothing about and given example of how easy it was to work out the biology of the region as much as GATA1.
The reason we bring this up is because we stared at our own tiling array data long enough to know how beautiful the known regions looked like. Troubles started, only when we tried to use that familiarity to make large-scale inferences about the entire genome.
-————————————————————–
On Evolution
ENCODE leaders have a very narrow view of evolution, which relies only on sequence conservation of a restrictive form. In their paper, they claim that evolutionary method (i.e. sequence conservation) is not enough to identify all ‘functional elements’ and therefore ENCODE type waste is necessary. However, that is complete nonsense. Evolutionary research works at many scales and here is a good example again from CRISPR. After the French group identified function of CRISPR repeats in one bacteria, other researchers observed the presence of similar sequence patterns in the genomes of a large number of bacteria and archaea. However, until the first discovery was made, most researchers analyzing those genomes ignored the same CRISPR blocks as useless.
The key point is that functionality can be inferred through evolutionary argument only by doing real biological experiments in many contexts/organisms and not by running as many large-scale experiments in human cells as possible. Now think about how many such real biological experiments could not be done due to ENCODE. Even if ENCODE’s $300 million were split into 300 $1M grants on random organisms and half of the groups did not find anything, we would have learned a lot more about functionality of human genes through evolutionary inference than what we learned from ENCODE.
-————————————————————–
Case for Abundant Junk DNA
The large section titled ‘case for abundant junk dna’ makes no evolutionary sense. Over the last three years, we have been working on a fish that is reasonably close to Danio (zebrafish) in evolutionary time-frame. The genome size of this fish is only half of zebrafish, even though it looks the same, grows the same and swims in the same way.
Yet this is what we see in our genomic data, when we make gene-by-gene comparison (there is considerable synteny). The exons are of nearly the same size in both fish species, whereas the introns are about half the size of Danio introns for a large number of genes and large number of intronic regions. We have made this comparison genome-wide and we have manually compared a number of genes for various reasons. We always found the introns to shrink by about 50%.
How can that happen, if 80% of Danio genome were functional? If at all, the fish we are working with should have more functional areas, because it has three big organs Danio does not have. With that understanding, check the following sentence from their paper -
ENCODE maps of polyadenylated and total RNA cover in total more than 75% of the genome. These already large fractions may be underestimates, as only a subset of cell states have been assayed.
Geez, apparently if you give these clowns more money, they can easily find evidence for 150% of human genome to be active. So, possibly the only solution is to give them 1/10th of money and get that 75% down to a healthy 7.5% :)
Joke apart, there is solid mathematics behind the above statement. Check -
Big Data Where Increasing Sample Size Adds More Errors
Conventional wisdom says larger sample size will make experiments more accurate, but that does not work in many situations. More samples can result in more errors, as explained by the following example.
Ask 1 million monkeys (~2^20) to predict the direction of the stock market for the year. At the end of the year, about 500K monkeys will be right and about 500K monkeys will be wrong. Remove the second group and redo the experiment for the second year. At the end of the year, you will be left with 250K monkeys, who correctly called the market for two years in a row. If you keep doing the same experiment for 20 years, you will be left with one monkey, who predicted the stock market correctly for 20 years in a row. Wow !
Or according to Taleb -
The winner-take-all effects in information space corresponds to more noise, less signal. In other words, the spurious dominates.
Information is convex to noise. The paradox is that increase in sample size magnifies the role of noise (or luck); it makes tail values even more extreme. There are some problems associated with big data and the increase of variable available for epidemiological and other empirical research.
Back to the ENCODE paper -
However, for multiple reasons discussed below, it remains unclear what proportion of these biochemically annotated regions serve specific functions.
Those ‘multiple reasons’ argue about anything from 5 percent to 5 gazillion percents, whereas throwing a dart is possibly a more exciting way to arrive at the answer. We expected the pilot ENCODE and ENCODE to answer those questions even if for a part of the genome, not sell us an expensive dartboard.
-————————————————————–
Lack of Null Hypothesis in Defining ‘Functional Element’
A few months back we covered an experiment of Mike White, which showed that “Random DNA Sequence Mimics #ENCODE !!”.
Last September, there was a wee bit of a media frenzy over the Phase 2 ENCODE publications. The big story was supposed to be that junk DNA is debunked ENCODE had allegedly shown that instead of being filled with genetic garbage, our genomes are stuffed to the rafters with functional DNA. In the backlash against this storyline, many of us pointed out that the problem with this claim is that it conflates biochemical and organismal definitions of function: ENCODE measured biochemical activities across the human genome, but those biochemical activities are not by themselves strong proof that any particular piece of DNA actually does something useful for us.
The claim that ENCODE results disprove junk DNA is wrong because, as I argued back in the fall, something crucial is missing: a null hypothesis. Without a null hypothesis, how do you know whether to be surprised that ENCODE found biochemical activities over most of the genome? What do you really expect non- functional DNA to look like?
In our paper in this weeks PNAS, we take a stab at answering this question with one of the largest sets of randomly generated DNA sequences ever included in an experimental test of function. We tested 1,300 randomly generated DNAs (more than 100 kb total) for regulatory activity. It turns out that most of those random DNA sequences are active. Conclusion: distinguishing function from non-function is very difficult.
Strangely, the new ENCODE paper completely overlooked that that important point. While ENCODE propaganda piece says on ‘What Fraction of the Human Genome Is Functional?’
Limitations of the genetic, evolutionary, and biochemical approaches conspire to make this seemingly simple question difficult to answer.
their lack of a null hypothesis should be treated as an even bigger ‘conspiracy’ !
-————————————————————–
Using Discredited GWAS Studies as Support for More Functional Blocks
ENCODE paper says -
Results of genome-wide association studies might also be interpreted as support for more pervasive genome function. At present, significantly associated loci explain only a small fraction of the estimated trait heritability, suggesting that a vast number of additional loci with smaller effects remain to be discovered. Furthermore, quantitative trait locus (QTL) studies have revealed thousands of genetic variants that influence gene expression and regulatory activity (9498). These observations raise the possibility that functional sequences encompass a larger proportion of the human genome than previously thought.
Give us a break, dudes !! ‘Thousands’ within a genome consisting of 3 billion nucleotides means nothing at all.
Moreover many of those same GWAS-type ‘discoveries’ are getting ENCODE-like treatment from respected scientists. For example, check what Bernard Strauss said about abundant false positives in newly discovered cancer mutations.
Mutation and Cancer A View from Retirement
What does seem clear is that the cancer genome project and the cancer atlas are examples of the inefficiency that is the consequence of funding large projects without accompanying large ideas. To be fair, given the impetus of the new technology it was probably impossible not to set these machines on to the available tumors in the expectation of finding druggable targets. However, the suggestion of the Lawrence paper that the ultimate solution will probably involve . . . massive amounts of whole genome sequencing amounts to a dogged adherence to a failed strategy similar to the massive attacks on the trenches by the Generals of World War I.
[snip]
The conclusion of the paper is that most of the mutations recognized as drivers are really false positives, the result of not having properly calculated control mutation rates.
-————————————————————–
Valuable Public Resource Excuse
The major contribution of ENCODE to date has been high-resolution, highly- reproducible maps of DNA segments with biochemical signatures associated with diverse molecular functions. We believe that this public resource is far more important than any interim estimate of the fraction of the human genome that is functional.
Q1. Who decided that those ‘public resources’ were needed?
Q2. Given that they were merely ‘resources’, did we get them at the best price achieved through competitive bidding?
-————————————————————–
Asymmetry of Press Releases
From another part of the paper -
Although ENCODE has expended considerable effort to ensure the reproducibility of detecting biochemical activity (99), it is not at all simple to establish what fraction of the biochemically annotated genome should be regarded as functional.
The entire paragraph and the subsequent one explains in great detail how difficult it is to call a transcript. In 2014, ENCODE leaders are telling us what those of us working on tiling arrays knew for long time, and we took great care in our 2003 paper not to over-inflate the results. Even in those days, we noticed the analysis by Affymetrix groups (led by Gingeras) to be on the side of making inflated claims, despite having lower quality of probes. What took them so long to learn?
Apart from that, we are puzzled that this PNAS paper making so many important and valuable points did not come with press releases and youtube videos, such as one from 2012.
-————————————————————–
‘Conflict of Interest’ Statement
The paper boldly states “The authors declare no conflict of interest”. We wish the authors simply omitted that statement, because it gives the impression that the authors are scientists giving unbiased and objective opinions. How about this as an example of ‘conflict of interest’ (from an ENCODE leader’s 2012 ENCODE press release)?
For the next phase of the ENCODE project, Weng received a four-year, $8 million grant from the NIH to lead the Data Analysis Center of the project.
It is clear to anyone who can think, that ENCODE leaders publish press releases with claims of making astounding discoveries and then get rewarded by Eric Green (an author of the paper) with more of other people’s money to waste.
Maybe it is time for scientists with interest in getting related large grants to mention any work on the project as ‘conflict of interest’ !
-————————————————————–
TLDR
Why this complete mockery of science called ENCODE does not get immediately shut down, when millions of real scientists are closing their labs, is far beyond our ability to fathom.