
Partitioning Libraries to Fit Small RAM Size
We keep harping on the issue of ‘too much sequence/too little RAM’, because it is the most frequent challenge faced by researchers working on assembling short read libraries. Today I received an email from a scientist trying to assemble three short read libraries. He said -
“I have 3 short insert size pe libraries and will have one long size pe (5k). I use abyss to assemble each library receptively because of the limited RAM.”
Is that the optimal strategy for partitioning sequence libraries?
We presented this figure in an earlier post.
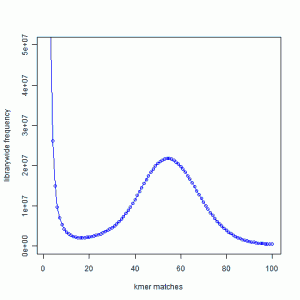
It shows frequency of 21-mers in a large read library, and is created in the following manner. We partitioned all sequences into contiguous 21-mers from both strands, and then counted those 21-mers for the entire library. Some 21-mers are present only once in the entire library, some are present twice, and some are present 50 or 100s of times. The peak around 50 comes from 21-mers that are actually present in the genome. It suggests that the genome is sampled about 50X by the collection of reads. The peak near 1 is the noise peak. For more details, please read the earlier post.
What happens, if we partition the library into three arbitrary sets and draw similar graph? The noise peak on the left will remain where it is, but the height of noise distribution will go down by about 1/3rd. The signal peak will shift to the left to around 16, because each partitioned library samples the genome 50/3=16 times. As a result, you will see a hyperbolic chart with two peaks merged into one. This is not good for the assembler, because it will have difficulty separating signal from noise.
A better strategy for partitioning would be to think in K-mer space instead of sequence space. We are not going to partition sequences, but partition pool of K-mers and let reads fall along.
i) Create a chart like above for the entire data set together.
ii) Find the K-mers that have frequency around 50, and then split those K-mers into groups.
iii) Partition the library based on K-mer groups.
It will not damage the K-mer distribution pattern, and therefore help you get better assembly for the same RAM.