
An Explanation of Velvet Parameter exp_cov
Appropriate choice of the ‘exp_cov’ (expected coverage) parameter in Velvet is very important to get an assembly right. In the following figure, we show data from a calculation on a set of reads taken from a 3Kb region of a genome, and reassembling them with varying exp_cov parameters. X-axis in the chart shows the exp_cov and y-axis shows the size of the largest scaffold assembled by Velvet.
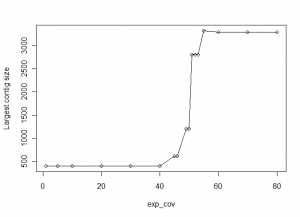
As you can see from the above figure, when exp_cov is low, Velvet has difficulty reassembling the original sequence. The largest contig assembled by Velvet is only 400 nucleotides long. As we increase the exp_cov, the size of largest contig gets bigger between exp_cov=50-60, which is indeed the coverage ratio for the reads in our library. For exp_cov above 60, the size of the largest assembled contig is close to the actual size of the original genomic sequence.
Why does the assembly change so dramatically with exp_cov parameter? To understand, you need to think about a large eukaryotic genome that has many repetitive regions.
Let us say a read differs from the genomic sequence at only one nucleotide position. There are two possibilities-
i) Unique region - The sequence, where the read comes from in the genome, is unique region. In that case, the read is ‘incorrect’ due to sequencing error or haplotype difference, and we can take a majority vote of many possible reads to determine the correct nucleotide at the mismatch position.
ii) Repeatitive region - The sequence, where the read comes from, is repetitive in the genome. Therefore, the difference in nucleotide is due to the read coming from another nearly identical segment of the genome.
Velvet does not know a priori whether (i) or (ii) is true for a read, because Velvet does not know the reference genome. In fact, Velvet’s job is to build up the reference genome, and so it has to make an intelligent guess about whether (i) or (ii) is correct for a read. The parameter exp_cov helps Velvet make that intelligent guess. Let us explain based on the above example.
Let us say the real coverage ratio is 55, but we tell Velvet that exp_cov is only 10. In that case, Velvet assumes that the reads varying slightly from each other are all coming from different repetitive regions of the genome. Therefore, Velvet attempts to assemble each of them separately.
On the other hand, if we tell Velvet that the expected coverage is 80, Velvet assumes that the reads varying slightly from each other all come from the same region of the genome. So, Velvet takes weighted average of varying reads to build up the region.
As you can see, it is wrong to give Velvet an exp_cov parameter that is too high or too low. If the number is given too low, Velvet comes up with a lousy assembly. If the number is given too high, Velvet merges all repeats into one large sequence. So, you may get a very high N50, but the underlying sequence can have many misassemblies. What should you do?
Option 1. You can use ‘-exp_cov auto’ and let Velvet guess the parameter. Please note that ‘-exp_cov auto’ is better than not giving any exp_cov to Velvet. In the above example, when we did not give any exp_cov to Velvet, it came up with the lousiest assembly, whereas when we gave ‘-exp_cov auto’, it chose exp_cov=60.9 and produced perfect assembly.
Option 2. You can estimate exp_cov yourself. Following two links may be of help.
BioStar: Should I be using k-mer coverage in Velvet parameters instead of nucleotide coverage?