
Genome Size Estimation and Polymorphism from K-mer Distribution
We were trying to understand the supplementary text on genome size estimation from BGI’s genome papers. Assembly experts Rayan Chikhi and BGI’s Zhenyu Li provided insightful answers that readers may find useful.
Rayan sent us two very informative links.
two good links are: http://seqanswers.com/forums/showthread.php?t=10988 and https://banana-slug.soe.ucsc.edu/bioinformatic_tools:jellyfish
Question to BGI:
I have been trying to follow the supplementary section of recent BGI papers (bat for example) and found the text on genome size estimation from k-mers. If I understand correctly, you split all reads into 17-mers, draw the distribution of how many distinct 17-mers are present one time, how many present 2 times, 3 times, etc. The peak of that graph gives depth of coverage, and you divide total amount of sequence with depth of coverage to estimate genome size.
I did a simulation on sea urchin genome 2 years back, and found that the peak itself shifts in a highly repetitive genome. Please check second plot below, where I sampled the genome 50X, but the peak appeared closer to 40.
http://www.homolog.us/blogs/2011/09/20/maximizing-utility-of-available-rams- in-k-mer-world/
If that is the case, how correct is your genome size estimation? I am asking, because your estimates seem too precisely defined and then you compare the assembled genome with that estimate to show that assembly was complete.
BGI - Zhenyu Li:
Hi,
I think you mixed the concepts of base coverage depth and kmer coverage depth.
When you refered 50X genome coverage, it meant base coverage which is obtained by
total_base_num/genome_size=(read_num*read_length)/genome_size.
Similarly, the kmer coverage depth, the peak value in kmer frequency curve, is calculated by
total_kmer_num/genome_size=read_num*(read_length-kmer_size+1)/genome_size.
So the relationship between base coverage depth and kmer coverage depth is:
kmer_coverage_depth = base_coverage_depth*(read_length- kmer_size+1)/read_length.
In your case, kmer_coverage_depth = 50 * (100 - 21 + 1)/100 = 40, which is exactly
the peak value in you plot.
best,
Question to BGI:
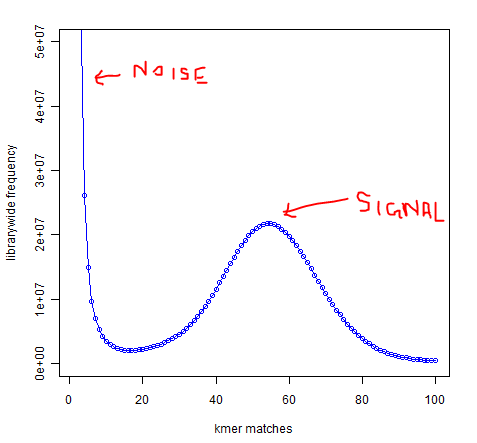
I have another question, if I may ask. When I draw distribution of distinct 21-mers present once, twice, etc. from an experimental DNAseq library, I see the first minima at 16 or 17, then the peak at 55. The peak at 55 represents coverage, but why does the first minima show up at 16-17? I would have expected the first minima (erroneous reads) to be around 2 -3, but 16/17 puzzles me. Do you have any experience with that? Is this shift happening due to high level of polymorphism in the genome?
Thanks a lot !!
BGI - Zhenyu Li:
I think the minima at 16/17 is mainly due to the sequencing error which is more serious than you expected.
And polymorphism usually causes a sub-peak at the position of half of the main peak
position (55 in your plot). Just like tha sub-peak caused by repeat kmer in you simulated data, where main peak is 40 and the sub-peak is 80, except that the position of sub-peak here is about 2-fold of the main peak. So I don’t think this minima at 16/17 is due to polymorhpism.