
Why is Gene Myers Dancing? Answer - Long Reads !!
A nice video came out of MPI Dresden, where you can find Gene Myers dancing holding his coffee cup in the hand.
The video had been going around in twitter for a couple of weeks, but nobody could explain what made him, his students and even creatures in the water so happy. Our NSA sources found out that it was the arrival of long reads. The biologists would not have to bear with the gappy zebra fish genome any more.
Speaking of assembling long PacBio reads, a few weeks back we reconstructed the algorithm of Gene Myers’ Dazzler assembler from the tweets of his talk -
Dazzler Assembler for PacBio Reads Gene Myers
PacBio enthusiasts will not have to rely on our third hand source any more, because Gene Myers started to release parts of his code and is also posting about it his personal blog -
Setup a blog for our DNA assembly work. Our data extraction tools are now available on github http://dazzlerblog.wordpress.com/2014/03/22/the- dextractor-module-save-disk-space-for-your-pacbio-projects/ . More to come.
The github page for Dextractor module is here.
The corresponding blog post can be accessed at the following link -
The Dextractor Module: Save disk space for your Pacbio projects
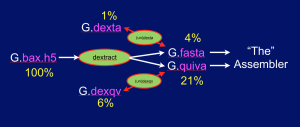
The Pacbio sequencers currently produce HDF5-formatted files that contain a lot of information about a sequencing run including the sequence, streams of quality values, and a lot of other stuff that most of us will never look at. These files are quite big. The 3 .bax.h5 files produced for a given SMRT cell typically occupy 7-9 Gb. The recent 54X human genome data set produced by the company consists of the output from 277 SMRT cells, so if you want to download it and work with it, you will need to set aside roughly 2+Tb of disk. Here we present a set of UNIX commands that extract just the data you need for an assembly project, and allow you to compress said data, so that it occupies roughly 1/13th the space. For example, the extracted and compressed human data set occupies only ~150Gb ! For our own projects we routinely extract and compress data as it is produced, saving the HDF5 files to a tape archive as backup. This saves our organization money as keeping such data sets on disk at these scales is not a cheap proposition.
The Dextractor module source code is available on Github here. Grabbing the code and uttering make should produce 5 programs: dextract, dexta, undexta, dexqv, and undexqv. Dextract takes .bax.h5 files as input and produces a .fasta file containing the sequences of all the reads, and a .quiva file that contains all the quality value (QV) streams needed by the Quiver program to produce a consensus sequence as the final phase of assembly. The .fasta file can then be compressed (~ 4x) with dexta to produce a .dexta file, and this can be decompressed with undexta to restore the original .fasta file. Similarly dexqv & undexqv compress (~ 3.3x) and decompress a .quiva file into a .dexqv file and back. The figure below shows the flow of information:
More details are available at his blog. Also check -
The Dextractor Module: Time and Space
