Trials and Tribulations with PacBio Data

While everyone reported their great success stories with PacBio data and hybrid assembly, we sat there with sad faces :(. What is the problem? Well, our PacBio data did not make sense at all.
Let us elaborate. We had a number of Illumina libraries on a fish genome and we assembled them with SOAPdenovo2 first. When we got our PacBio data with ~1.2X coverage, we aligned those on the assembled genome and found a bunch of long reads not to match the genome at all.
That is not a big deal, because SOAPdenovo2 and other de Bruijn graph-based assemblers do not cover the repetitive regions well. So, it is quite possible that PacBios were giving us better resolution in the repetitive parts of the genome.
Well, there was one problem, or two to be more precise.
i) The number of long PacBio reads not matching the assembled genome was too big. They suggested that our assembly did not cover ~30% of the genome. We did a lousy assembly !!
ii) We are not afraid to announce that we did a lousy genome assembly, but here is the second problem. No matter how hard we tried, we could not prove that the SOAPdenovo2 assembly was bad. We ran Minia with every imaginable k-mer length. We ran SGA. We even went to the extent of finding every single 21-mer from Illuminas and trying to match with the missing PacBio reads. No matter what, those reads could not be accounted for.
Chen-Shan (Jason) Chin of PacBio was of great help. He suggested that we run BLASR of those PacBio reads on other PacBio reads and see whether they match with other ones. It was because the PacBio sequencing protocol is far more unbiased than Illumina protocol and can sequence regions of the genome that Illumina cannot. So, if the PacBio reads were real, they would have some matches against each other. We did that found that indeed those PacBio reads had matches with other reads. So far so good. Still we were puzzled why the Illumina machine missed so many regions of the genome.
This morning, we were playing with a sorted file containing information on PacBios that aligned with the genome and those that did not, and noticed something odd. In the file, the reads matching genome were clustered together and the reads not matching genome were clustered together. That means the file had twenty lines on reads matching the genome and then five lines of reads not matching the genome. Another twenty lines of reads matching the genome and so on. What the hell is going on??? The reads were sorted based on their IDs. How did the PacBio machine know so much and gave the matching and non-matching reads such IDs that they clustered together at the end of of bioinformatics?
The answer turned out to be quite simple. Along with 10 SMRT cells of real data, our sequencing center packed 3 SMRT cells of data from some other organism. No wonder they were not matching our genome. After we excluded those 3 SMRTs, we have >95% match and very good overlap with the scaffolds of existing assembly.
The only silver lining is that during this whole process, we read every imaginable PacBio-related paper and tried every possible algorithm to find a clue.
Have a nice weekend !
-——————————————————
Reader @happy_khan asks, whether we could have detected the error by aligning unassembled reads against NR database.
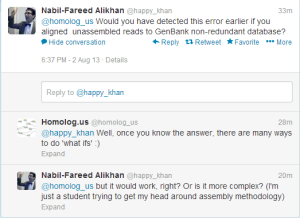
Maybe and maybe not. Here is why.
The NR database contains the protein-coding genes and not the entire genomes. We do not know, what those 3 SMRT cells contain, but if they have the genome of another organism, only a small part could have aligned with NR. That, of course, depends on what the other organism is. If it were E. coli, whose genome is packed with genes, NR search could have possibly given many hits. If the other sample were human, there is only 1 in 10 chance of hitting coding regions within NR.
Second problem - the primary mode of error in PacBio reads is indel and BLAST does not work with them very well. So, we have to run BLASR against NR to find matches. That is not how all online databases for running NR search are set up. So, one needs to download the NR genes and run BLASR locally to find out what is going on.
Even if NR contained entire genomes of all sequenced organisms, we could not have gotten many hits if those 3 SMRT cells had a new organism. Outside coding regions, the genomes change quite a bit from one organism to another.
We will run NR search some time to figure out what those samples are, but are too tired for today.
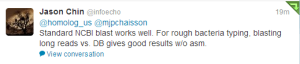
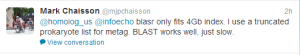
Edit. It seems like BLAST has no problem with PacBio. Thanks to Deanna Church for the helpful comment.
Jason Chin and Mark agrees.