
Is BWA-MEM as Good as BLASR for Aligning PacBio Reads? – Part 2
In an earlier commentary, we asked whether BWA-MEM is as good as BLASR for aligning PacBio reads. A number of readers, including the authors of both programs, commented here and in twitter. Below we present some preliminary results from our comparison. Please excuse us, if the following commentary appears too text-heavy. We are still in the middle of the analysis and will add a few graphs as we continue.
An Introduction to PacBio Data Format
For this comparison, we used a subset of human genomic reads released by PacBio and Eichler lab covering the genome at 10X. For those unfamiliar with PacBio data, here is a quick introduction.
PacBio sequencing is done in SMRT cells containing zero-mode waveguides (see video at the link). As a bioinformatician, all you need to know is that the data from each SMRT cell comes with an identifier like -‘m120131_103014_sidney_c100278822550000001523007907041295_s1_p0’ or ‘m130610_155918_42210_c100539712550000001823089611241300_s1_p0’. The ID is likely designed by a committee and figuring out what it means is beyond your pay grade :). However, it is enough to know that the part ‘c100539702550000001823089611241314’ represents an unique identifier for the SMRT cell, and the part before that reflects the mood of the sequencer in some way.
The read libraries come in fasta, fastq or h5 format. The native format for PacBio is bax.h5, whereas fasta or fastq are derived from the h5 files. Each SMRT folder usually contains three files splitting all sequenced reads.
i) m130929_161837_42213_c100518541910000001823079209281315_s1_p0.1.fasta,
ii) m130929_161837_42213_c100518541910000001823079209281315_s1_p0.2.fasta,
iii) m130929_161837_42213_c100518541910000001823079209281315_s1_p0.3.fasta.
Within each file, the reads are shown as -
m130929_161837_42213_c100518541910000001823079209281315_s1_p0/1
AAGTTGT…….
m130929_161837_42213_c100518541910000001823079209281315_s1_p0/2
TCTGCTGGATCGTCAAGAGGTAAAAACGCA…..
and so on. The numbers after “/” go from 1 to say 55K in file m130929_161837_42213_c100518541910000001823079209281315_s1_p0.1, ~55K to ~110K for m130929_161837_42213_c100518541910000001823079209281315_s1_p0.2 and ~110K to ~165K in m130929_161837_42213_c100518541910000001823079209281315_s1_p0.3.
As a first step of the analysis, you will need to filter and clean the raw reads and, most importantly, split them into subreads. After that preprocessing step (‘pls2fasta’ in BLASR library), you will get a second set of files, with names like -
i) m130929_161837_42213_c100518541910000001823079209281315_s1_p0.1.subreads.fa sta,
ii) m130929_161837_42213_c100518541910000001823079209281315_s1_p0.2.subreads.f asta,
iii) m130929_161837_42213_c100518541910000001823079209281315_s1_p0.3.subreads. fasta.
The subreads FASTA file will contain IDs such as -
m130929_161837_42213_c100518541910000001823079209281315_s1_p0/163432/27907_34 462
m130929_161837_42213_c100518541910000001823079209281315_s1_p0/163432/34510_36 038
That means the sequence marked as “m130929_161837_42213_c100518541910000001823079209281315_s1_p0/163432” in original file got split into two, and the splitting locations are given as the extra parameter on the right. Some reads from the original file will disappear at this point and some other reads will get included at full length.
An example from human libraries
For our analysis, we chose data from m130929_161837_42213_c100518541910000001823079209281315_s1_p0 SMRT cell. It is one of those 66 SMRT cell covering the human genome at 10X, and therefore likely contains 10/66 or 15% of the genome. The actual number for this SMRT cell is slightly lower - ~389M nucleotides covering the genome at 12%.
Speaking of number of reads, the original raw read files had at least 163,465 reads (we are guessing, because that file is not released). Among them, 110K reads disappeared after the pre-processing and the remaining 53,933 reads got split into 62,617 subreads. Do not worry about the great loss of 110K reads. Preprocessing step usually sees 15-20% loss of nucleotides constituting of mostly small or adapter sequences. Therefore, it is quite impressive that, with the new chemistry, PacBio achieved 500Mb/SMRT cell of throughput after pre-processing and not with the raw reads.
The N50 read size for this SMRT cell is 10,092 or ~10K after pre-processing. The longest subread has size of 41,463 nt and about 1,500 subreads are longer than 20Kb each.
BWA-MEM vs BLASR
Both Heng Li (author of BWA MEM) and Mark Chaisson (author of BLASR) commented on the applicability of BWA MEM on PacBio reads in the earlier thread. Based on our understanding of the algorithms, we believe Mark’s comment is more accurate and here is why. When someone says that PacBio reads contain 15% of errors, people tend to imagine 1 error or more in every 10 nucleotides. Mark mentioned a few months back that he often puts up two slides in his conference presentations - one with roughly 1 error in every 10 nucleotides and another with an actual PacBio read (15% error rate) - both aligned to some reference. When he asks the audience to identify the real PacBio read, they almost always select the incorrect one.
In reality, PacBio reads have longer stretches of matching segments than what our naive expectation would suggest. That is not a special feature of PacBio technology, but is an outcome of the mathematics of random numbers. Truly random coin tosses have much longer stretches of all heads or all tails than what our naive expectation of 50-50 probability distribution would suggest. BLASR paper uses this observation to find large matching seeds between the query and the reference, and then uses collection of matching seeds to generate the overall alignment.
BWA-MEM is no different except for one difference. BLASR paper by Chaisson et al. found that optimal seed size for PacBio reads would be ~16nt, whereas BWA- MEM sets the default seed size at 19nt. Apart from that, the seeding strategy of BLASR and BWA-MEM appear to be similar.
Speed, Memory, Performance
Why care about BWA-MEM, when BLASR is already available? The answer is speed. BWA-MEM is about 8-10 times as fast as BLASR. For the SMRT cell mentioned above, BWA-MEM took one hour to align the entire library, whereas BLASR is estimated to take ~8 hours (based on aligning 1/8th of the reads in one hour). For both alignments, we used default parameters.
We do not know, which algorithmic step leads to the largest difference in speed. Most likely, it is from the two-way BWT used by BWA-MEM. Please note that we ran BLASR after pre-computing the suffix array for the genome and providing it as an input in alignment step. Therefore, suffix array generation time is not included in the time difference between the algorithms.
Number of Matches for 53,933 Reads in the above file
We split the matches into four categories.
(i) BWA mem and BLASR match the read to the same chromosomal region:
46,952 reads (87%) in an estimate highly biased toward BWA MEM (see later).
(ii) BWA mem and BLASR map the read to different chromosomes:
2266 reads (4%).
(iii) BWA mem can map the read, but BLASR cannot:
3987 reads (7%).
(iv) BLASR can map the read, but BWA mem cannot:
728 reads (1.3%).
Let us take a closer look at each category to see what is going on. First group counts agreement between two algorithms, but what is ‘agreement’? We are counting all cases, where BWA MEM and BLASR map the read on the same human chromosome, but the starting location of the map may vary. The following chart shows the number of matches by varying the distances between starting locations of BWA MEM and BLASR map. The 46,952 number reported earlier allows distance of as much as 10K betweeen BWA MEM and BLASR hit (has to be same chromosome), but with 5K distance, the number of agreements is still at a respectable 46,336.
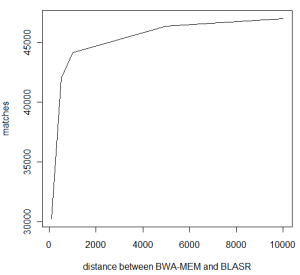
Is 5Kb distance too large to say that BWA MEM and BLASR ‘agree’? Apparently not. We went through the individual alignments, and find that often BWA MEM maps part of a read in the right location, but does not extend the match to both ends. For longer reads, BWA MEM leaves out 3Kb or more of unmapped parts on each side, whereas BLASR forces end-to-end map, as commented by Mark Chaisson. It is possible to extend the BWA MEM match by dropping the gap penalty to 0? We have seen that to work, but have not done any systematic analysis yet. A number of readers also commented in twitter BWA MEM does not go over long gaps well. What we have seen is that BWA MEM splits such matches into two separate hits, but once again, no systematic analysis has been done yet.
Second category - “BWA mem and BLASR map the read to different chromosomes”. We manually checked a few such cases and found that BWA MEM often returned the correct map identified by BLASR as its second best hit. This needs to be further investigated.
Third category - “BWA MEM can map the read, but BLASR cannot”. We have no insight into this group. It is possible that BWA MEM aligned only a small fragment (50-100 nt, possibly repeat) of the read, whereas BLASR, by forcing end-to-end match, could not see any proper agreement.
Fourth category - “BLASR can map the read, but BWA MEM cannot”. We followed up on a few such hits and found that their percentage differences with the reference, as reported by BLASR, were around 76-77%. Maybe the seeding strategy of BWA MEM fails, when the error level is too high.
We will continue to work on this topic, and report any other miracle or divine intervention that occurs to us.