
New Paper and Raw Data: "A first look at the Oxford Nanopore MinION Sequencer"
Here is the definite guide to Nanopore that you are looking for. Reader Alexander S. Mikheyev kindly emailed us from Japan with his paper on evaluating nanopore sequencer.
1. Download raw data and the paper from his github page
Evaluating the performace of Oxford Nanopore’s MinION
Citaton: Mikheyev, A.S. and Tin, M.M.Y., A first look at the Oxford Nanopore MinION sequencer, Molecular Ecology Resources, in press.
The complete contents of the data folder will eventually be found on the [Dryad Digital Repository]
Currently the data folder includes the raw data in FASTQ format. If you need any other files, please contact the authors.
2. Library Prep
They used two library prep kits from Oxford Nanopore to evaluate
the sequence the lambda phage genome (genomic DNA kit) and cDNA from the venom gland of the pitviper, Protobothrops flavoviridis (amplicon kit). The runs were performed on separate flow cells. Quoting from the paper -
Library preparation is similar to that for other next-generation applications, requiring DNA shearing, end repair, adaptor ligation and size selection. Once these steps are complete, the library must be conditioned, and then it is ready to load on the sequencer. Library preparation takes about half a day, and is of
comparable complexity and cost to library preparation for other platforms. Finally DNA is conditioned by the addition of a motor protein, a step that requires a 30-min incubation that can be extended to overnight, for a greater number of 2D calls. The libraries are then mixed with buffer and a fuel mix, and loaded
directly into the sequencer.
3. Cost of sequencing
The flow-cell survives for 48 hours. Based on that and throughput given by Loman, you can compute the cost. However, take a look at point 5 to estimate true cost.
For optimal yield, the MinKNOW must be re-filled with additional library every four hours during a 48 hour run, which is the lifetime of a flow cell.
4. Bioinformatics tools
For all libraries, reads were mapped to the reference using BLASR, a mapper developed for long, relatively inaccurate reads of the Pacific Biosciences sequencer (Chaisson & Tesler 2012), and also using BLASTN (Altschul et al. 1990) (Table 1).
5. Read length, yield and accuracy
Although the raw yield of the MinION is impressive, both in terms of read lengths and data output, the accuracy was extremely low. For the lambda phage, about 10% of the reads actually mapped to the reference using BLASR; their identities were 2.2% and 8.9% for the 1D and 2D workflows, respectively. This means that much less than 1% of all the generated sequence faithfully matches the reference. BLASTN produced similar results, although more short sequences were detected, resulting in a higher percentage of mapped reads, but not leading to greater overall coverage (Table 1). The major sources of error in MinION data were indels, particularly insertions that introduce spurious data (Figure 3B).
The paper has lot more information including their results on snake cDNA that you do not want to miss. Here is a hint - “Results were far worse for the snake cDNA library amplicon sequencing run”.
Overall, nanopore appears to be a promising technology and we expect the quality of sequences to improve over time.
-—————————-
Edit.
Nick Loman disagrees. Truth is on Loman’s side? Mikheyev’s side? In the middle?
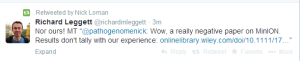
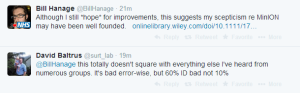
Others think the number of matches is in 60% range, not 10%.
Announcement.
Nick Loman and Oxford Nanopore CEO Clive Brown are going to host a Google hangout conference on Sept 10th to discuss nanopore data. No flying needed ! Anyone can attend from any place in the world.
Wed, 10 Sep, 06:00 - 09:00
Hangouts On Air Broadcast for free
The popular Balti and Bioinformatics series returns, this time in virtual space! Join us for a nanopore-themed programme which will be streamed from Google Hangouts to YouTube.
Draft schedule (subject to change)
Times are British Summer Time (GMT+1)
Start: 14:00 BST, 13:00 GMT, 09:00 Eastern Standard Time
14.00 - Intro
14.05 - Clive Brown, Nanopore sequencing
14.45 - Nick Loman, Early data from nanopore sequencing: bioinformatics opportunities and challenges
15.15 - Matt Loose, Streaming data solutions for nanopore
15.30 - Josh Quick, Nanopore sequencing in outbreaks
15.45 - Torsten Seemann, Awesome pipelines for microbial genomics
16.15 - Finish