
Nanopore - Truth and Nothing but the Truth
At last we get the analysis of Oxford Nanopore data that we had been looking for since first day. Michael Schatz posted the GI2014 slides of James Gurtowski from his lab in his website.
-—————————-
Read Length Distribution
Read length goes all the way to ~147Kb, but the longer reads are problematic (see next section).
Mean read length is 5.4Kb.
-—————————–
Aligned Reads
Only 32% of the reads match anywhere in the genome. Long reads have high failure rate. Here is the relevant slide.
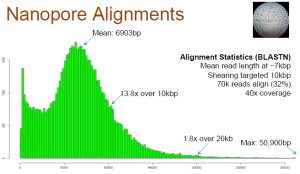
Also note that the reads, which match, may only align partially (~50%). Check slide 10.
-—————————-
Error rate
For reads that match, average accuracy is 64%. That number increases to 70% for the ‘2D base calling’ reads. These are much better than the initial report of Mikheyev/Tin and that is definitely good news. Moreover, another poster presentation at the conference reported similar numbers.
Distribution of errors - 57% Mismatches, 32% Deletions, 11% Insertions.
Please note that the distribution is quite different from PacBio, which is mostly dominated by ~70% insertions.
-—————————-
Implication for Assembly and Other Work
1. One thing not clear from the slides is whether the errors are distributed randomly within reads or have bias. This is important for ‘perfect assembly math’ presented by Gene Myers in his AGBT talk.
2. What is the probability that a random base from a 40KB read is actually from the genome? We know that the read is 32% likely from the genome and within the read, 50% match the genome on average. Also, the rate of accuracy for matching reads is 70%. So, the probability is 0.320.50.7=11%.
The middle number will be much better for shorter reads and worse for the long reads.
3. With 32% matching rate and 65-70% accuracy for matching regions, it will be impossible to do assembly from Nanopore reads alone. This will have implications for doing field work. The assemblies in the presentation were done by combining Nanopore and Illumina reads.
An important point in this respect -

-—————————————-
A week back, we mentioned about Nick Loman’s release of E. coli data at Gigascience website. Readers will find the following arxiv paper helpful (h/t: @lexnederbragt).
Background: The MinION is a new, portable single-molecule sequencer developed by Oxford Nanopore Technologies. It measures four inches in length and is powered from the USB 3.0 port of a laptop computer. By measuring the change in current produced when DNA strands translocate through and interact with a charged protein nanopore the device is able to deduce the underlying nucleotide sequence. Findings: We present a read dataset from whole-genome shotgun sequencing of the model organism Escherichia coli K-12 substr. MG1655 generated on a MinION device during the early-access MinION Access Program (MAP). Two sequencing runs of the MinION were performed using the R7 chemistry (released July 2014) and one using R7.3 (released September 2014). Conclusions: Base-called sequence data are provided to demonstrate the nature of data produced by the MinION platform and to encourage the development of customised methods for alignment, consensus and variant calling, de novo assembly and scaffolding. FAST5 files containing event data within the HDF5 container format are provided to assist with the development of improved base- calling methods. Datasets are provided through the GigaDB database http://gigadb.org/dataset/100102 Keywords: genomics; nanopore sequencing