
A Sequel to Heng Li’s Mysterious New Program
One month back, our blog alerted the readers about a new program ‘bfc’ being developed by Heng Li. Check - Heng Lis Mysterious New Program.
This week, Heng Li posted a preprint and, as we suspected, it is indeed an error-correction program.
Correcting Illumina sequencing errors for human data
Summary: We present a new tool to correct sequencing errors in Illumina data produced from high-coverage whole-genome shotgun resequencing. It uses a non- greedy algorithm and shows comparable performance and higher accuracy in an evaluation on real human data. This evaluation has the most complete collection of high-performance error correctors so far.
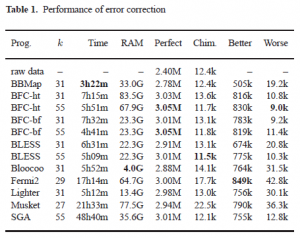
Heng Li wrote a detailed blog post explaining where it stand w.r.t. other error correction programs.
When I started to evaluate BFC, I was only aware of SGA, Lighter and the old BLESS. Lighter-1.0.4 is very slow on compressed input. Old BLESS does not work as the test data have reads of variable lengths, while new BLESS is so challenging to build that I gave it up initially. I was quite happy to see BFC was several times faster than these tools.
I was overoptimistic. When I monitored the Lighter run more closely, I found it was mostly single-threaded. I then tried Lighter on uncompressed input. It was six times as fast as the run on compressed input. I told the Lighter developers my findings and suggested solutions. They were very responsive and quickly improved the performance. The timing reported in the preprint was from the new Lighter.
Having seen Lighter could be much faster, I decided to get BLESS compiled. It was a right decision. The new BLESS is working well, especially when it uses long k-mers. This has inspired further explorations: use of KMC2 and long k-mers for error correction. I implemented a variant of BFC, BFC-bf, to take KMC2 counts as input and keep the counts in a bloom filter. I have to ignore base quality during the counting phase as KMC2 is not aware of base quality in the way I would prefer. The correction accuracy is not as good as BFC. Nonetheless, BFC-bf helps to confirm that the apparently better correction accuracy is not purely due to the use of base quality during k-mer counting. BFC-bf also makes it easier to use long k-mers.
The role of k-mer length in error correction is complicated. Since assembling corrected reads takes much more computing time and my development time, I have not run de novo assembly often at the beginning. I assumed that low under- and over-correction rates should lead to better assemblies. I was wrong again. At least for fermi2, short k-mers used for correction lead to better assemblies (for reasons, see the discussions in the preprint). I have also tried to combine two k-mer lengths but only with limited success.
Here I should admit that a weakness of the manuscript is that I have not run de novo assemblies for all correctors. This is mostly because I do not have enough computing resources. Another key reasons is the assembly result varies with assemblers. It is too much for me to try M assemblers on N correctors. I did briefly try fermi2 assembler on E. coli data corrected by a few (not all) tools. BFC is better, which is a relief but not conclusive. I need to try more assemblers on more data sets to get a firm view. This is for future.