
E. coli Genome Assembled using Nanopore Data Only
Finally -
A complete bacterial genome assembled de novo using only nanopore sequencing data
A method for de novo assembly of data from the Oxford Nanopore MinION instrument is presented which is able to reconstruct the sequence of an entire bacterial chromosome in a single contig. Initially, overlaps between nanopore reads are detected. Reads are then subjected to one or more rounds of error correction by a multiple alignment process employing partial order graphs. After correction, reads are assembled using the Celera assembler. We show that this method is able to assemble nanopore reads from Escherichia coli K-12 MG1655 into a single contig of length 4.6Mb permitting a full reconstruction of gene order. The resulting assembly has 98.4% nucleotide identity compared to the finished reference genome.
Github repos are available here and here.
They are using DALIGNER by Gene Myers, as we suggested some six months back. Wondering why HGAP pipeline did not work, given that it would have been the easiest solution.
Details of the method -
(i) Four Minion runs were used and only the 2D reads were considered. Average read length - 4kb-8kb. Accuracy - 78-85%.
“In total, 22,270 2D reads were used comprising 133.6Mb of read data, representing 29x theoretical coverage of the 4.6 megabase E. coli K-12 MG1655 reference genome.”
(ii) Assembly method - DALIGNER –> multiple rounds of POA (correction tool using similar method as pbdagcon) –> Celera assembler.
POA is referenced to - “Lee, C., Grasso, C. & Sharlow, M. F. Multiple sequence alignment using partial order graphs. Bioinformatics 18, 452464 (2002).”
(iii) Entire genome was captured in one contig.
Overall, a clean and well-written paper.
Also coming -
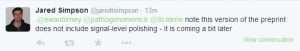
-————————
Criticisms of the assembly paper started appearing.
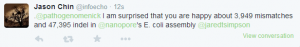
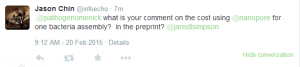
Grab your popcorn. It is going to be fun !!