
Pacbio Talks at #AGBT - What to Expect
About one and half year back, I wrote two-part blog posts starting with the following comment. The full posts are linked below.
I will go out on a limb and make a bold call. The world of genomics is on the verge of seeing another set of major transformations, and many algorithms, tools, pipelines and methodologies developed for short reads over the last 3-4 years will be useless. In my opinion, the era of short-read sequencing is reaching a peak, or to be kind to its users, short read technologies are shining like the full moon. Related to peaking of the short read era, we will see two other changes (i) end of genome sequencing and genome paper era and (ii) end of big data bioinformatics. For further explanation of the last sentence, please read the detailed explanation in the later part of the commentary.
End of Short-Read Era? (Part I)
End of Short-Read Era? (Part II)
Nobody believed me at that time, but things are changing in small ways and big ways. As an anecdote of small change, a friend of mine showed the reviews of his recent proposal, where he requested funds for genome assembly using Illumina PE and mate-pair reads. The reviewer asked him to get rid of mate pairs and use Pacbio reads. Speaking of big change, you probably have noticed the words ‘perfect assembly’ in the title of Gene Myers’ talk.
On this developing story, here is what I expect to hear from the talks at Pacbio.
Algorithms
Two battles had been going on in the algorithmic front, and both of them can be described as ‘Gene Myers vs Gene Myers’.
The first one is on alignment, where the goal was to take BWT and SA out to save time from BLASR. The DALIGNER built by Myers solved that problem.
In DALIGN Paper, Gene Myers Delivers a Major Blow to His Biggest Competitor
The second goal is to take Celera assembler out and go to a more direct approach for building string graph and assembly. Jason Chin’s Falcon does that, and I expect Myers to announce a similar algorithm in his talk.
Applications
What is the best application of long reads that can place it way ahead of various short read technologies? I display a few random tweets from AGBT to show you where the short people are stuck.
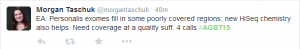
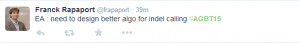
As you can see, they are so bogged down with finding indels and solving coverage issues that developing algorithms to find the sequences of two independent chromosomes would take a long time. The words ‘diploid’ or ‘highly polymorphic’ rarely enter the short read bioinformatics literature. So, in a masterful stroke, Pacbio is focusing on pulling out two copies of the chromosomes and a disease where the information matters.
Oxford Nanopore - are they going to compete?
Previously, I have been critical of Oxford Nanopore, and the main drawback, as I argued, was the absence of someone showing a nanopore-only genome assembly. As I discussed, the entire premise of carrying USB stick-sized sequencer falls apart, if you have to also carry a MiSEQ along. Now that Jared Simpson and collaborators completed E. coli assembly from nanopore data only, the previous objection does not remain valid. So, that is definitely a major step ahead IMHO.
Now the question of which technology is better will be decided based on quality+cost, with portability being an added benefit of nanopore sequencing. Will servers win in the long run or iphones? Will servers win in the long run or palm-pilots? As you know, SGI servers are out of business, Dell servers are hot, Iphone has been a winner and palm-pilots can be found in junkyards. So, do not buy/sell stocks based on anything you hear in this blog. I can only discuss where things stand in the technological front.