RNAseq Annotation Issues - A Good Post by Stuart Brown
In “Pig Genome Problems”, Stuart Brown wrote:
I have had a few issues with genome annotation in the past couple of weeks that highlight how dependent NGS methods are on these reference files. In a mouse RNA-seq project, we found that RNA reads from some olfactory receptor genes were getting low expression counts (using TopHat/Cufflinks) because RNA reads were not mapping within the known exons of the genes. When we looked at the actual alignment BAM files in IGV, it was clear that the exon boundaries in the reference (RefSeq) genes were wrong. Both the 3’ and 5’ ends of some genes were missing regions with lots of RNA alignments. It would be nice to make a new reference genome using our RNA-seq reads to create our own annotation, but it is actually quite difficult to get one RNA sample with all genes expressed at useful levels.
We put the ‘olfactory receptor genes’ in bold, because those receptor genes stay in clusters and cause major problems in assembly and annotations, especially from short reads. His remaining commentary is quite informative on everyday issues being faced by bioinformaticians, and he ends with -
My only conclusion from these issues with reference genomes is that we depend on these things to be right, and when they are not, NGS data analysis can be badly messed up. There is no simple solution for providing a perfect reference for every genome for every purpose, so bioinformaticians need to stay on their toes and dig deeper when results from standard methods don’t make sense.
The process of going from sequencing to new discoveries can be broadly summarized in the following steps.
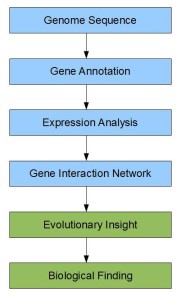
The steps colored in blue were noisy even before cheap short-read sequencing and short reads exacerbate the problems. Using evolutionary filters can be one way to reduce the noise, long reads being the other. Whatever it is, reducing noise adds to costs and often becomes the dominant cost in the process. So, we chuckled, when we saw the following ad at the end of his commentary: