
The Mathematics of Color Space Sequencing
‘Next-generation’ sequencing technologies are dramatically transforming medical fields and life sciences. ABI SOLiD machines are very popular, because they can sequence an order of magnitude more nucleotides than most other competing machines (454, Solexa, etc.) for the same cost. However, bioinformaticians analyzing SOLiD data have to confront the puzzling world of color space. Hopefully, the following discussion will reduce some of the confusions. We will only tackle the mathematics behind color space sequencing here, and leave the chemistry of SOLiD sequencing for another day.
Let us say that we are interested in sequencing a DNA segment. A typical sequencing machine will sequentially identify every nucleotide as A, C, G, T and report a long stream of ATGGTGGTTGTTACTGCGCGTGGGAACCCCCTG… etc. Instead, the SOLiD machine reports transitions between neighboring nucleotide pairs.When one looks at the pairs of neighboring nucleotides, the number of reportable combinations increase from 4 (A, C, G, T) to 16 (AA, AC, AG, AT, CA, CC, CG, CT, GA, GC, GG, GT, TA, TC, TG, TT). To simplify reporting, the SOLiD machines elegantly reduce the possible combinations from 16 to 4based on the following table:
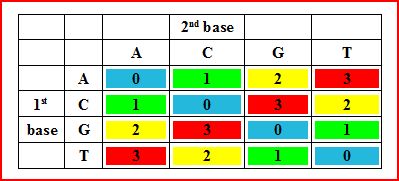
You see that four combinations - AA, CC, GG, TT - are all reported as 0. Another four combinations - AC, CA, GT, TG - are reported as 1, and so on.
We shall soon elaborate on what kind of thought went behind choosing the numbers in the above table.Also we shall show that this reduction of complexity comes at a cost. First, let us explain how the color space works.
####How to convert sequences to color space?
Let us choose a specific example (ATGGTGGTTGTTA). The sequence will be converted in the following manner -
A
-
T
-
G
-
G
-
T
-
G
-
G
-
T
-
T
-
G
-
T
-
T
-
A
-
3
1
0
1
1
0
1
0
1
1
0
3
In the color code table shown earlier, the first transition from A-T is noted as red or 3. The second transition from T-G is noted as green or 1. Continuing in the same manner, the entire sequence ATGGTGGTTGTTA will be converted to
- That is the color space representation of the sequence.
If we choose another example - GCAACAACCACCG, we soon discover that it also converts to 310110101103. Two other sequences - CGTTGTTGGTGGC and TACCACCAACAAT also have the same representation.Do you see the problem here? The nucleotide space sequence of a color space representation is not unique. Every color code data can be converted to nucleotide space in exactly four different ways. This is due to reduction of complexity from 16 to 4, while choosing the numbers in color code table.
To avoid ambiguity, SOLiD machines report the first nucleotide along with the remaining color space representation. Therefore, the four sequence will be given as A310110101103, C310110101103, G310110101103 and T310110101103. Once thefirst nucleotide is known, color space data can be converted to nucleotide space in an unique manner.However, we will soon see that this conversion back to nucleotide space is inefficient and eliminate the primary advantage of SOLiD sequencing. Analysis of color space data needs to be done in color space.
How do we compute reverse complement in color space?
The reverse complement of the original sequence (ATGGTGGTTGTTA) is (TAACAACCTCCAT). Here we convert it into color space.
T
-
A
-
A
-
C
-
A
-
A
-
C
-
C
-
T
-
C
-
C
-
A
-
T
-
3
0
1
1
0
1
0
1
1
0
1
3
You can see that this color space representation is the exact opposite of the original color space data (310110101103).This is true for all sequences. That is how the numbers in the conversion table were chosen.
Simple sequences
Low complexity sequences also show two or four-fold degeneracy in color space. For example,
000000…..
AAAAAA…. or CCCCC…. or TTTTTTT…. or GGGGGG….
111111…..
ACACACA…. or GTGTGTGT…..
222222…..
AGAGAG…. or CTCTCT……
333333…..
ATATATAT…. or CGCGCGCG…..
SNP
If we introduce one nucleotide SNP in the original sequence, its color space representation gets two changes. You can see that by comparing the following example with the original sequence. The modified region is marked in red.
A
-
T
-
G
-
G
-
T
-
G
-
G
-
A
-
T
-
G
-
T
-
T
-
A
-
3
1
0
1
1
0
2
3
1
1
0
3
One base change in the color space, on the other hand, can dramatically change its nucleotide space representation. For example, the sequence A310110201103 with one color space difference from the original 310110101103 translates back toATGGTGGAACAAT. Do you see how different it is from the original ATGGTGGTTGTTA.
We provide two Perl scripts to convert any other sequences from color space to nucleotide space or from nucleotide space to color space.
Advantage of color space
The last example shows the greatest advantage of color space sequencing. Sequencing machines are error prone. Among all errors, single nucleotide changes are most common. This is a problem for all resequencing projects, because their primary goal is to identify single nucleotide changes from a reference sequence. Oftenit is not clear whether the single nucleotide change observed after sequencing is due to sequencing error, or whether it is a genuine difference from the reference. When oneworks with color space sequencing, true SNPs can be easily distinguished from sequencing errors. A real SNP marks two changes in color space from the reference genome, whereas a sequencing error does not translate to anything close to reference genome.
We note that color space data from SOLiD machines often contain single errors. If one converts all sequences to nucleotide space, the converted data do not look anything remotely close to real sequence and therefore all subsequent analysis may become highly error prone. Therefore, it is advisable to perform as much computational analysis in color space as possible. It is better to convert the reference data to color space than SOLiD sequences to nucleotide space.
Disadvantage
The above constraint becomes the primary disadvantage of color space data. Over the years, many analysis tools had been written for nucleotide space sequences. They often do not work in the color space, and moreover, at the end of the day, we have to get data converted to nucleotide space and it may not always be possible. For example, one can perform de novo assembly of an unsequenced region in color space, but how does he get the real sequence of the unsequenced region?
Pseudo basespace
The first disadvantage can be overcome by using a concept called ‘pseudo basespace’. A large number of software tools were written for nucleotide sequences. Let us say you have a color space sequence that you like to align with a reference sequence using Clustal.As we discussed before, converting color space SOLiD sequence into nucleotide space is not optimal. The solution is to convert the reference genome to color space, and then replace (0=A, 1=C, 2=G, 3=T) in both converted reference sequence and SOLiD sequence. This tricks Clustal into aligning two sequences. After alignment, one can make reverse conversion (A=0, C=1, G=2, T=3).
One must be cautious in using the pseudobase space conversion. For example, if one uses BLAST with pseudobase space converted data, the reverse complement matches given by BLAST cannot be trusted. This is because reverse complement of SOLiD sequences are done differently from what would be done for true nucleotide space sequences.
Our follow up posts on this topic -
Do de Bruijn Assemblers Work in Color Space?
Trinity and Contrail for Color Space