
End of Short-Read Era? - (Part I)
I will go out on a limb and make a bold call. The world of genomics is on the verge of seeing another set of major transformations, and many algorithms, tools, pipelines and methodologies developed for short reads over the last 3-4 years will be useless. In my opinion, the era of short-read sequencing is reaching a peak, or to be kind to its users, short read technologies are shining like the full moon. Related to peaking of the short read era, we will see two other changes - (i) end of “genome sequencing and genome paper” era and (ii) end of big data bioinformatics. For further explanation of the last sentence, please read the detailed explanation in the later part of the commentary.
I spent the last two days in California attending an user group meeting of Pacific Biosciences and am absolutely fascinated by the talks and discussions. You may say that the views expressed here are colored by many PacBio only talks and that is true to some extent. However, I went into the meeting with a healthy dose of skepticism, even though I heard many positive opinions on PacBio technology for months. I know Dr. Jason Chin, the author of HGAP paper and a leading bioinformatician at PacBio, for many years. He is a very smart ex-physicist and was my collaborator in two important papers. Over the last eighteen months, he had been telling me that I should abandon trying to assemble genomes from short reads. Despite knowing him for long time, I assumed that he was biased and was trying to oversell his company’s technology. Now I am embarrassed, because his description of the technology appears to be quite accurate, if not a modest description of the range of possibilities.
Cost per Base is a Superficial Measure. Cost per Information Content is More ‘Informative’
Few weeks back, Sergey Koren posted a paper titled - Reducing assembly complexity of microbial genomes with single-molecule sequencing in the arxiv. The paper discussed how PacBio helped in finishing a number of bacterial genomes.
BioMickWatson responded with - Bacterial genomes 2nd and 3rd generation costs and came to the conclusion -
My conservative estimate is that PacBio is about 10 times more expensive per sample for bacterial genomes than Illumina, and in reality it is probably higher.
His comment about expensive versus cheap boiled down to how many nucleotides one can get for a fixed amount of money, which I believe is a superficial way of seeing things. Think about it this way. We know that PacBio reads are 85% accurate. Therefore, only 85 out of 100 nucleotides of PacBio reads are informative. In the zeroth order, that should make the PacBio reads 15% less useful than a simple nucleotide cost comparison would suggest.
But why stop at the zeroth order? In the first order, we know that PacBio reads are about two orders of magnitude longer than the Illumina reads. Positional data provided by long reads is another form of information. In this comparison PacBio wins, especially after RS II and BluePippin size selection. So, we get two orders of magnitude of information gain with PacBio for one order of magnitude of extra cost as estimated by BioMickWatson.
Then again, others would argue that Illumina mate pair reads provide positional information too, and that point is not lost on BGI or the authors of the SPAdes assembler. BGI, for example, recently published the tiger genome paper, where they pushed the mate pair insert size to 20 kb.
Libraries for the Amur tiger genome were constructed at BGI, Shenzhen, and the insert sizes of the libraries were 170?bp, 500?bp, 800?bp, 2?kb, 5?kb, 10?kb and 20?kb.
We also need to factor in the bioinformatician’s time needed to scaffold longer mate pairs. The assembly process is not fully automated and can add errors here and there (Assemblathon 2 anyone?). Sorting out those errors increases the cost of assembly, which a pure sequencing cost does not include.
Taking all those points into account, it is not at all easy to estimate the cost per information content, but that does not mean we need to resort to superficial measures like cost per nucleotide.
Genome Assembly
Is there any reasonable way to compare cost per information content for two technologies? I would argue that complete genome assembly is one such problem. Complete genome is a type of information that biologists consider as important. Both Illumina and PacBio technologies are attempting to provide that information through two different routes.
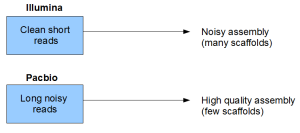
You can see that even though short reads are clean themselves, the read size itself is a form of noise or loss of information. That loss of information manifests itself into the assembly quality of the genome.
How about relative costs? Alex Copeland of JGI gave a fantastic talk, where he discussed how the assembly costs and qualities of microbial genomes changed over the years.
2002-2006 - $50K/genome, 49 contigs (Sanger)
2006-2008 - $35K/genome, 22 contigs (Sanger)
2008-2011 - $10K/genome, 44 contigs (Sanger)
2011-2013 - $1.5K-$3K/genome, 69 contigs (switch to Illumina)
2011-2013 - $5K/genome, 6 contigs (PacBio)
Based on his numbers, the switch to PacBio increased his costs by about a factor of 2, but led to big improvement in the quality of the assembly. In the microbial world, PacBio has become a winner.
Then I heard the same about the fungal genomes, where JGI managed to assemble complete chromosomes with PacBio. Chongyuan Luo of Salk Institute presented on high quality assembly of various Arabidopsis strains (Col-0, Ler-0, cvi) from PacBio only. HGAP was quite successful there as well (access data here) . Given that Arabidopsis genome is 120 Mb long, I do not see why assembling smaller vertebrate genomes from PacBio only would pose any difficulty. HGAP paper was published only this year and nobody attempted the method on larger genomes only due to lack of time.
There will be no Assemblathon 3
As I explained earlier, short read sequences are clean themselves, but have severe loss of information through read sizes. That loss of information manifests itself into the genome assembly. Assemblathon 1 and 2 were the responses of genomics community to estimate that ‘noise’ in genome assembly.
Given that large PacBio sequences already removed the assembly noise from genomes as large as Arabidopsis, I do not see what the purpose of Assemblathon 3 would be. Will Assemblathon 3 be restricted to short reads only to quantify the relative success rates of short read-related algorithms in removing noise? Will it include very difficult assembly problems that only PacBio reads can handle? I suspect Assemblathon 3 will face operational difficulties, because it will not only have to evaluate assembly algorithms but will also have to compare different sequencing technologies.
However, when it comes to PacBio only, their Arabidopsis work already made extensive comparisons between the existing Arabidopsis assembly and the new assembly to show that PacBio produced high quality assembly. Not only that, the comparisons led to understanding of why the assemblies were incorrect in some regions and further refinement of the assembly algorithm. After those corrections are accounted for, there is little need to worry about assembly error. Hence no Assemblathon 3.
End of Genome Sequencing Era
PacBio makes assembly so easy that there will be no glory in genome assembly. The genome sequencing era started in the mid-90s with the publication of genome papers of various model organisms, but reached massive media frenzy in 2000-2001, when human genome papers were published. Biologists are generally rewarded for publishing in Science and Nature and genome papers had been the surest way to get there.
In the first generation, genome papers used to be 100 pages long and journals devoted entire issues for the supporting papers. Then the journals started to shrink space devoted to genome papers and pushed much of the material to supplement. Supporting papers were asked to go elsewhere.
In the late stage, genome papers turned into a very predictive meme -
(i) X is a cool organism,
(ii) We assembled high-quality genome of X, which will help in figuring out why X is cool.
(iii) Title: “Genome of X reveals why X is cool”, where ‘reveal’ refers to few genes identified through genome comparison and some straightforward bioinformatics.
Essentially the genome papers got into Nature or Science for picking a cool organism and completing (ii). With long reads from PacBio making step (ii) easy, I do not see why genome papers should get any importance for merely completing the engineering task of assembling high-quality genomes. We also noticed that a paper comparing the genomes of white Bengal tiger, African lion, white African lion and snow leopard did not make into a glam journal. So, possibly BGI saturated the field even prior to arrival of PacBio.
Assembly of Difficult Regions - Major Histocompatibility Complex
What is biologically relevant information? So far we talked about assembly of entire genomes and argued that higher quality genome assembly is more informative than cleaner short reads. However, many scientists do not benefit from the entire genome unless the genomic regions they are working on are assembled properly. Given that NIH spends a large part of its budget to cure human diseases, one can say that immune genes in mammals is an informative piece of the puzzle contributing to the research of many biologists.
Stanford immunologist Lisbeth Guethlein presented on -“Genomic Architecture of the KIR and MHC-B and -C Regions in Orangutan” and explained how PacBio helped them immensely in reconstructing those relevant genes.
If you are unfamiliar with MHC, here is a short introduction from wiki -
The major histocompatibility complex (MHC) is a set of cell surface molecules encoded by a large gene family in all vertebrates. MHC molecules mediate interactions of leukocytes, also called white blood cells (WBCs), which are immune cells, with other leukocytes or body cells. MHC determines compatibility of donors for organ transplant as well as one’s susceptibility to an autoimmune disease via crossreacting immunization. In humans, MHC is also called human leukocyte antigen (HLA).
….
The MHC gene family is divided into three subgroups: class I, class II and class III. Diversity of antigen presentation, mediated by MHC classes I and II, is attained in multiple ways: (1) the MHC’s genetic encoding is polygenic, (2) MHC genes are highly polymorphic and have many variants, (3) several MHC genes are expressed from both inherited alleles.
The Stanford group tried to assemble the MHC region of Orangutan for many years, first using Sanger and then from 454 sequences. Then came PacBio and they got their results within days without spending any time on bioinformatics. That was quite fascinating, because I am stuck with very similar assembly difficulties for smell and odor receptors in another organism. Receptor genes sit in clusters and are very similar to each other, making their assembly from short reads extremely difficult. Before going to the meeting, I have been working on an algorithm to sort out tandem duplicated genes from de Bruijn graphs. Now I wonder whether that is at all necessary and whether I should spend my time elsewhere.
Assembly of Diploid and Polymorphic Genomes
Now that the problem of getting high-quality genomes from next-generation sequencing is solved, researchers can move on to other difficult problems such as phasing. Separating diploid genomes from short reads is very difficult, especially when the chromosomes differ by a large extent (>5%). BGI had hell of a problem with the oyster genome and they came up with an innovative method that required extensive resequencing of shorter segments of the genome. Long PacBio reads would make the process quite easy. Chongyuan Luo from Salk Institute mentioned that they were working on mixing reads from two separate strains of Arabidopsis to see whether the strains can be computationally separated out.
Transcriptome Assembly
PacBio developed a terrific transcriptome assembler - ‘CRAZY’. It assembles the transcripts and isoforms easily and to a high degree of accuracy. Bye bye Trinity !!
If you are wondering about it, ‘CRAZY’ is not an acronym. One has to be crazy to think about assembling transcripts from PacBio reads, given that the reads themselves are longer than typical genes. You sequence and you get your genes. No assembly needed.
Gene Expression Measurement
Transcriptome experiments have two components - (i) finding the genes, (ii) determining relative gene expressions. Illumina is a clear winner on the counting aspect.
We worked on transcriptomes for over a decade and find the introduction of PacBio to lead to another major shift as shown below.
i) First generation: Genes were determined through gene prediction programs and some EST. Expressions were measured through spotted arrays.
ii) Second generation: Genes were determined through EST, comparative analysis and later tiling arrays. Expressions were measured using oligonucleotide arrays.
iii) Third generation: Short read technologies helped in both determining the genes and estimating relative expressions.
iv) Fourth generation (future): Genes will be determined through PacBio experiment, and relative expression will be measured through short read sequencing.
Finding Methylation Patterns and other Genome Modification
Matthew Blow from JGI gave an excellent presentation on bacterial functional genomics. PacBio is the only sequencing technology capable finding genome modification directly from the same signals for identifying nucleotides. The talk was quite informative on the biological aspects and I will see whether I can find the slides to post here.
Part II of this commentary covers metagenomics, pricing aspects, what the changes mean to biologists and core facilities, and whether Pacbio can survive as a business to make the above changes happen.