
Sharing a K-mer Story
We started with 600 million 100mer reads from a genomic library and plotted its K-mer distribution. It looked like the following plot.
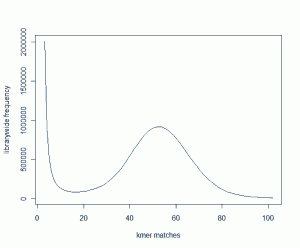
The peak around 52 suggests that non-repetitive regions of the genome are covered ~52 times by the reads. For details about the basic philosophy of what is being plotted and why, please read our earlier commentary on Maximizing Utility of Available RAMs in K-mer World.
When we added another 800 million 51mer reads into the pot, the K-mer distribution changed to the following image. The peak of two combined libraries moved to 58.
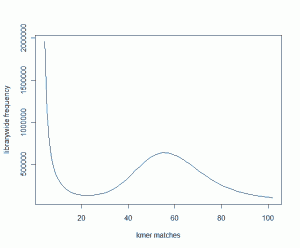
Does that make sense? The new library has about 70% as many sequences as the old one. Shouldn’t the peak shift from 52 to 88?
Stay tuned for an explanation.