An Opinionated History of Genome Assembly Algorithms - (ii)
Dear readers, We wanted to devote this commentary to the history of genome assembly algorithms from 1995 to today, as promised in An Opinionated History of Genome Assembly Algorithms - (i). However, when we tried to qualify the sentence “if a Nobel prize is ever awarded for the human genome, he (Gene Myers) should get it alone”, it was impossible to restrict the discussion to computer-science concepts only. At the end, we decided to split the commentary into two parts - this one on the ‘biological’ aspects of the human genome project and the next one on the technical developments. Remember, everything is ‘opinionated’ and some commentaries are more opinionated than others :).
-—————————————————————————-

1988 Video of Eric Lander talking about Human Genome Project (click on image to listen) - his ‘functionator’ was finally ‘demonstrated’ by Ewan Birney in 2012 ENCODE paper
-—————————————————————————-
Human Genome Project Put on Five Year Plans
Anyone growing up in the former communist world laughs hearing the words ‘five year plans’, because those plans typical symbolize utter incompetence. Chinese had their five year plans during Mao’s time, Soviets had them during Stalin and the most incompetent government of all continues to develop ‘five year plans’ today. In contrast, economic conditions in China improved dramatically, when its government had the good sense to ignore its ‘plans’.
Apparently, NIH did not get the memo, when it placed the human genome project on five year plans. In all fairness, it was not entirely of NIH’s fault. When the project was conceived (1988), USSR was still going strong and all 54 of 50 ‘Soviet experts’ in Wasington DC saw the system to last for ever.
Nevertheless, the human genome project itself was a giant boondoggle that had been off to a rocky start. The original plan was to finish the project in 2005 after three five year plans. To convince the US public that money was not being wasted, a reputed scientist like James Watson was placed as the project leader. The story-line fascinated the public - sixty year old James Watson, who discovered DNA structure in 1953, is now heading the NIH project to decode the human genome and staking his reputation to make sure that money is not being wasted.
Once the public money was secured, keeping Watson was too much of an inconvenience and the opportunity to fire him came soon. A ‘patent crisis’ emerged in 1991 as we covered in an earlier commentary.
AT A CONGRESSIONAL BRIEFING ON THE Human Genome Project last summer, molecular biologist Craig Venter of the National Institute of Neurological Disorders and Stroke dropped a bombshell whose repercussions are still reverberating throughout the genome community. While describing his new project to sequence partially every gene active in the human brain, Venter casually mentioned that his employer, the National Institutes of Health, was planning to file patent applications on 1000 of these sequences a month.
I almost fell off my chair, says one briefing participant who asked not to be named. James Watson, who directs the genome project at NIH, did more than that, exploding and denouncing the plan as sheer lunacy. With the advent of automated sequencing machines, virtually any monkey can do what Venters group is doing, said Watson, who in one sentence managed to insult Venter, his dismayed postdocs, and Reid Adler, the director of NIHs Office of Technology Transfer, who advised Venter to pursue the patents. What is important is interpreting the sequence, insisted Watson. If these random bits of sequences can be patented, he said, I am horrified.
Watson was gone in 1992 (check Watson Departure Vexes Genome Experts and Why Watson Quit as Project Head) and the new head Francis Collins joined in a year (1993). The changes created some minor inconveniences for the planners. By the time Collins boarded as captain, the ‘five year plans’ were off target by three whole years. Therefore, 2003 was selected as the new ending year (1993 + 2 five year periods). It was the perfect year for a new PR story - human genome will be decoded in the 50 year anniversary of publication of Watson/Crick paper.
Francis Collins turned out to be the perfect leader for the boondooggle as well. By his third year as leader of HGP, he retracted five of his own papers and put the entire blame on a junior collaborator (‘trainee’) for falsifying data. One of those papers had only two authors - Collins and the ‘trainee’. So, clearly Collins never bothered to critically think about a scientific paper before putting his name on it and relied entirely on the other colleague. That theme of senior researchers taking all credits for good papers but no blame for bad/fake papers emerged as a major model of NIH- sponsored science in Collins’ time.
-—————————————————————————-
A Quick Recap on Stalin’s Five Year Plans, If You Are Feeling Nostalgic
Redears are warned not to not fall for the ‘success’ of the first five years. It was achieved through ‘holodomor’ (extermination by hunger) of Ukrainians, which took place in 1932-33.
The Holodomor (Ukrainian: ?????????, “Extermination by hunger” or “Hunger- extermination”;[2] derived from ‘?????? ???????’, “Killing by Starvation” [3][4][5]) was a man-made famine in the Ukrainian Soviet Socialist Republic in 1932 and 1933 that killed up to 7.5 million Ukrainians.[6] During the famine, which is also known as the “Terror-Famine in Ukraine” and “Famine-Genocide in Ukraine”,[7][8][9] millions of citizens of Ukrainian SSR, the majority of whom were Ukrainians, died of starvation in a peacetime catastrophe unprecedented in the history of Ukraine.[10] Since 2006, the Holodomor has been recognized by the independent Ukraine and several other countries as a genocide of the Ukrainian people.
-—————————————————————————-
1998 Update of Five Year Plans
In 1998, Francis Collins and crews published their first report in a Science paper and projected what would happen in the next ‘five-year’ period.
New Goals for the U.S. Human Genome Project: 1998-2003
The paper, authored by NHGRI head Francis Collins, DOE head Ari Patrinos, Elke Jordan, Aravinda Chakravarti, Raymond Gesteland, LeRoy Walters and other central planners, was full of newspeak such as “an ambitious schedule has been set to complete the full sequence by the end of 2003, 2 years ahead of previous projections” (emphasis ours). In reality, the numbers 2003 and 2005 had little to do with actual work and were derived from other political considerations (explained earlier). Collins’ paper contained other apparently ‘bold’ forecasts, such as the ability to finish fly genome by 2002, mouse genome by 2005 and other mammalian genomes by the end of the 21st century (Note: we made the last one up, but it is not far from reality). The larger strategy remained to be clone-by-clone sequencing, which was slow and labor- intensive.
If you are thinking that the planners were doing public a favor by reducing the time-line of five year plans from 15 years to 10 years, you are wrong. In another ‘policy’ paper published in Science earlier, Collins, Mark S. Guyer and Aravinda Chakravarti started to pitch for their next boondoggle - the human HapMap project.
Variations on a Theme: Cataloging Human DNA Sequence Variation
New methods for the discovery and scoring of single-nucleotide polymorphisms (SNPs) offer the potential for considerably improved methods for genetic analysis of complex biological phenomena, particularly common diseases. In this Policy Forum, the authors call for a publicly supported effort to discover a large number of SNPs and to place the information in public databases. Participation in this public effort by the private sector would be particularly desirable.
Their simplistic understanding of human diseases was challenged by Andrew Clark, Ken Weiss, Deborah Nickerson and colleagues, but that did not result in any change of heart.
Many of our findings may seem to be inconsistent with the success of a number of investigators in the identification of genes for disease phenotypes in populations founded relatively recently by a small number of individuals. Examples are the Amish population, in which Hirschprung disease has been studied (Chakravarti 1996), and studies of North Karelia populations, in which a number of diseases have been mapped successfully (Peltonen 1996). Such successes have led to the view that a dense SNP mapbeing even more definitive, in terms of a simpler underlying mutational process would be useful in an even broader population context (e.g., the United States). The explanation for this apparent inconsistency seems to be straightforward. First, the disease phenotypes mapped in this way, to date, usually are rare, often are of single- locus etiology, and exhibit clear Mendelian inheritance in families. In a small population, such traits are likely to have only a small number of causal alleles, so that samples ascertained by such phenotypes are likely to comprise clones of descendants of the founding allele(s).
-—————————————————————————-

Aravinda Chakravarti - “Not everything we eat is curry” and not every way we fall sick is Mendelian
-—————————————————————————-
Hypothetical Case of Little Johnny
In the meanwhile, Francis Collins started to sell another story-line to the public and this one also had very specific years and timelines. In a paper published in 1999 in New England Journal of Medicine, he wrote -
A HYPOTHETICAL CASE IN 2010
General visions of gene-based medicine in the future are useful, but many health care providers are probably still puzzled by how it will affect the daily practice of medicine in a primary care setting. A hypothetical clinical encounter in 2010 is described here.
John, a 23-year-old college graduate, is referred to his physician because a serum cholesterol level of 255 mg per deciliter was detected in the course of a medical examination required for employment. He is in good health but has smoked one pack of cigarettes per day for six years. Aided by an interactive computer program that takes John’s family history, his physician notes that there is a strong paternal history of myocardial infarction and that John’s father died at the age of 48 years.
To obtain more precise information about his risks of contracting coronary artery disease and other illnesses in the future, John agrees to consider a battery of genetic tests that are available in 2010. After working through an interactive computer program that explains the benefits and risks of such tests, John agrees (and signs informed consent) to undergo 15 genetic tests that provide risk information for illnesses for which preventive strategies are available. He decides against an additional 10 tests involving disorders for which no clinically validated preventive interventions are yet available.
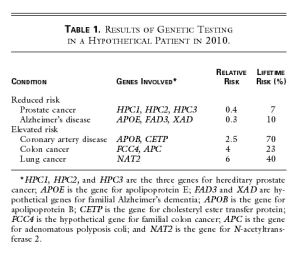
A cheek-swab DNA specimen is sent off for testing, and the results are returned in one week (Table 1). John’s subsequent counseling session with the physician and a genetic nurse specialist focuses on the conditions for which his risk differs substantially (by a factor of more than two) from that of the general population. Like most patients, John is interested in both his relative risk and his absolute risk.
John is pleased to learn that genetic testing does not always give bad news his risks of contracting prostate cancer and Alzheimer’s disease are reduced, because he carries low-risk variants of the several genes known in 2010 to contribute to these illnesses. But John is sobered by the evidence of his increased risks of contracting coronary artery disease, colon cancer, and lung cancer. Confronted with the reality of his own genetic data, he arrives at that crucial teachable moment when a lifelong change in health-related behavior, focused on reducing specific risks, is possible. And there is much to offer. By 2010, the field of pharmacogenomics has blossomed, and a prophylactic drug regimen based on the knowledge of John’s personal genetic data can be precisely prescribed to reduce his cholesterol level and the risk of coronary artery disease to normal levels. His risk of colon cancer can be addressed by beginning a program of annual colonoscopy at the age of 45, which in his situation is a very cost-effective way to avoid colon cancer. His substantial risk of contracting lung cancer provides the key motivation for him to join a support group of persons at genetically high risk for serious complications of smoking, and he successfully kicks the habit.
The ‘Table 1’ from the paper (Johnny’s report from genetic test) is shown below -
That paper was clearly misleading and Collins should have known that it was, if the 1988 talk by Lander (linked above) or real evidence by Clark et al. were of any indication. Lander ended with -
But the Human Genome Project, per se, will tell us none of that. But it will be the tool which we can use in conjunction with a powerful functionator as we together try to build one over the next decade or so to answer most of those questions.
Also, note that those forecasts about personalized medicine were made along with the 1998 projection of sequencing timeline, i.e. human genome would be sequenced by 2003, mouse by 2005 and a number of other mammalian genomes over the following decades. Therefore, Francis Collins had no clue, what would unfold in technological terms over the next 10+ years (454, Illumina, PacBio, etc.) and foresaw a ‘personalized healthcare’ world with only one human genome, one mouse genome and little bit more. In reality, he had little clue of what would unfold in the next 10+ months.
-—————————————————————————-

Francis Collins - was so busy retracting papers that he never listened to the ‘functionator’ part of Lander’s talk
-—————————————————————————-
Eugene Myers Crashes the Party
Getting back to the Science paper by the planners, in addition to ‘bold’ projections about sequencing timelines, it also called Gene Myers a crank in technical language (“major concerns”).
A whole-genome shotgun strategy has been proposed previously [J. Weber and E. W. Myers, Genome Res. 7, 401, but major concerns have been raised (P. Green, ibid., p. 410), about the difficulties expected in obtaining correct long-range contig assemblies. It will not be possible to evaluate the feasibility, impact, or quality of the product of this approach until more data are available, which is not estimated to occur for about 12 to 18 months. See also R. Waterston, J. E. Sulston, Science 287, 53 (1998).
What did Weber and Myers do wrong? In a Genome Research paper published earlier that year, they had the audacity to propose a radically different method that would be way faster and cheaper than clone-by-clone sequencing to finish the human and other genomes. In their approach, bulk of the work would be done by a smart computer program (now known as ‘shotgun assembler’) working to join together read fragments sequenced randomly from the entire genome.

Government-backed HGP scientists did not like that message at all, because it created several inconveniences.
(i) Reducing the cost of genome sequencing 10-fold meant reducing the power of the bureaucrats by 10-fold; not a happy prospect, when the money being spent was ‘other people’s money’.
(ii) Human genome project had to be finished in 2003, not a day sooner or later, or the ‘five year plan’ would be discredited.
It is noteworthy that Stalin’s five year plans also started to face severe difficulties by the second five year period.
Unfortunately, most government-backed HGP scientists did not have a proper response. By that time, Gene Myers was in his seventh year after Kececioglu’s PhD and had all tools developed to back his claims. The most relevant piece among them was a powerful simulation program that could account for many different error rates and repeat conditions, and show that his assembly method could scale nicely for large genomes. Simulation was the only mean to argue, since there were no real sequences from the human genome except for the small pilot study (1% of genome).
Phil Green, a reputed computational biologist, wrote a meticulous rebuttal - “Against a Whole- Genome?Shotgun”. A rejection by Phil Green was no small matter in those days. Like Waterman, he authored an important paper with Lander about the human genome assembly (check Construction of multilocus genetic linkage maps in humans), but more importantly, he was the only person, whose assembly-related programs (PHRED, PHRAP and CONSED) were being widely used. However, it will be hard to believe that his judgement was not clouded by the source of his funding - National Human Genome Research Institute and the Department of Energy. For example, if Phil Green wrote a meticulously written rebuttal of hyped up claims by Francis Collins of the benefits of human genome sequencing or overuse of Mendelian genetics, we have not seen it.
Another criticism came from Evan Eichler (Masquerading Repeats: Paralogous Pitfalls of the Human Genome), where he made passionate case against Myers’ ideas based on his experience with duplicated genes with clinical consequence.
The critics would argue that these regions represent junk DNA (a term borne out of ignorance and not necessarily a biological property of our genome) and therefore should not be a priority for sequencing.
….
Let us not short-change ourselves and future generations of scientists by opting for the cheapest and quickest route in the generation of a complete human genomic reference sequence.
In 1999, Myers tried in vain to address those complaints in a RECOMB paper written with Eric Anson. There they showed how it would be possible to connect distant contigs by using BAC-end sequences, because inability to get long- range assembly was the most serious criticism. The RECOMB paper was generally ignored and received only 36 citations. By then, there was another reason to ignore him: he left academia and joined a sequencing company.
-—————————————————————————-
Celera Published Drosophila Genome
HGP central planners nearly had heart attack in March-April 2000, when Gene Myers and Celera surprised the world with two announcements. First, they published the full assembly and analysis of the Drosophila genome, two years before the second five year plan. That was not a simulation, but the assembly of a real genome published in the most prestigious science journal. Second, they announced that they were nearly done with sequencing human genome and would start sequencing mouse in a week. Assembly was a nearly automated step in their whole-genome shotgun approach.
Media picked up the story and declared Celera to be the winner of ‘human genome race’. That meant the publicly funded monstrosity would be questioned by the public.
Celera Genomics has finished sequencing the entire human genome.
The private company made the surprise announcement Thursday morning at a House hearing that had been scheduled to discuss the future of the Human Genome Project.
“We’ve finished the sequencing phase,” Celera president Craig Venter said at the hearing.
With those words, Celera officially beat the public Human Genome Project in a long, closely watched race that ended several months ahead of Celera’s own schedule as well as the public project’s.
“It’s awesome. It’s an incredible scientific feat. They really pushed the envelope of the technology and they’ve done what nobody thought could be done … well ahead of schedule - it’s remarkable,” said Cyrus Harmon, president and CEO of Neomorphic, a genomics company in Berkeley.
The next step, Venter said, is to assemble the mapping annotation over the next several weeks: Celera scientists will piece the fragments of the sequence together according to where they’re located in the body.
-—————————————————————————-
Who Won the Human Genome Race?
The public project was put on a fast-track because of Celera’s announcement, and both Celera and public genome projects published their human genome papers in early 2001. Subsequently, a number of papers were published by HGP scientists with the claim that Gene Myers and Celera did not deliver and HGP scientists were right all along.
For example, check -
Whole-genome disassembly - Phil Green
On the sequencing of the human genome - Waterstone, Lander, Sulston
We do not want to call any ‘winner’ of the ‘race’, because not only the ‘race’ was not properly defined but a race with public money is not worth running. Almost every article written about Celera versus public HGP seem to be biased in one way or another. Some of them try to sell ‘private companies are more efficient’ angle and some others try to argue that ‘Celera used public data in combination of its own data, because open access is great’ angle. Give us your favorite social hypothesis and let us see whether we can create a ‘Celera vs public HGP’ story out of it :).
-—————————————————————————-
Contribution of Weber/Myers in Retrospect
Instead of asking who won the human genome race, if, instead, we evaluate the merits of 1998 suggestions by Weber/Myers, we find that those suggestions impacted biology/medicine in two profound ways.
(a) Shotgun Assembly Approach for Large Genomes
In 1998, very few scientists accepted the notion that it would be possible to assemble large complex genomes in an automated manner using whole-genome shotgun method. That attitude changed after the demonstration of Drosophila assembly by Celera. Later, MIT/Whitehead Institute developed its ARACHNE assembler and other genome centers followed with their own methods. Those algorithmic developments will be discussed in the following commentary. Whole- genome shotgun turned out to be the method of choice in genome assembly, because the other alternatives were prohibitively expensive.
(b)Genome Assembly is a ‘Software Release’, not Publication of a Paper
The second contribution of Weber/Myers was in the manner of doing things. The HGP scientists considered the genome assembly similar to the final publication of a paper, and expected all i-s to be dotted and t-s to be crossed. The product had to be of the highest quality (complete genome 99.99% accuracy and all possible disease regions covered).
Instead, Weber/Myers presented genome project as an engineering problem, where three parameters were optimized - cost, quality, speed of release. They wrote -
We assert that the goals listed in Table 1 are the true motivation for sequencing the human genome, not the accomplishment of some arbitrary, mythical goal of 99.99% accuracy of a single, artifactual (in places) and nonrepresentative copy of the genome. Most research laboratories, both public and private, want discrete genomic sequence information, and they want it as early as possible. They are interested in information such as the intron/exon structure of specific genes, the polymorphisms that may occur in specific coding and regulatory sequences, and lists of coding sequences that lie within specific chromosomal intervals. The sooner this critical information is available, the sooner it can be applied to accelerating research progress. Americans spend ?$35 billion per year, public and private, on biomedical research (Silverstein et al. 1995). If the efficiency of this research is improved by even 1%, and this is probably a gross underestimate, then savings would be $350 million per year, far more than the cost of sequencing. Whole- genome shotgun sequencing will allow these savings to be realized far sooner than with clone-by-clone sequencing. We should generate as much of the critical sequence information as rapidly as possible and leave cleanup of gaps and problematic regions for future years.
It is not too late to change strategies for sequencing the human genome. Only a few percent of the sequence has been generated at this time. Even if the human genome is not sequenced via the shotgun approach, there are still many other large genomes that will be sequenced in the future, including many agriculturally important species. It will likely be too expensive to sequence other large genomes via the clone-by-clone approach. A possible general strategy for sequencing other large genomes would be a random cDNA sequencing project, followed possibly by some radiation hybrid physical mapping of the ESTs, followed by whole-genome shotgunning.
The above text was a profound change from the thinking of the scientists involved with HGP, because it proposed the genome assembly to be similar to ‘software release’. A programmer starts with releasing an ‘alpha version’ of his program, which is crappy for most users, but useful for some dare-devil early users. The subsequent ‘beta version’ is useful for more users, and but may contain flaws. Conservative users need to wait for v1.x release. Weber and Myers correctly foresaw the need for early release within the community.
Over the following decade, the leaders of almost all other genome projects tilted in favor of getting fast and inexpensive near-complete draft genomes, when the other alternative (very high quality finished genome) turned out to be expensive. Genome completion surely has value, but only if it can be obtained in an automated manner or through advanced technology (check “A Sunset for Draft Genomes?”).
For an example of change of heart based on evidence, read the following recent comment by Jonathan Eisen, who previously argued in favor of getting a complete genome in 2002 - (The Value of Complete Microbial Genome Sequencing (You Get What You Pay For) - Claire M. Fraser, Jonathan A. Eisen, Karen E. Nelson, Ian T. Paulsen, and Steven L. Salzberg).