
A Question on MaSuRCA Assembler
We wrote about MaSuRCA assembler a few months back. Today a reader asked the following question by email -
I was reading the paper as well as your blog on MaSuRCA assembler and I have a simple question. If they choose the unique k-mer from k-mer counting table for read extension, then isn’t that k-mer is erroneous? The k-mer counting tools always get rid of k-mers with low frequency, right?
Answer is simple as well :). As Anton Korobeynikov commented in the previous thread, MaSuRCA is building a de Bruijn graph under the hood. That means they are splitting the reads into k-mers, but storing the k-mers in a table instead of RAM. Once you have that, you can proceed along the de Bruijn graph one base at a time and join the neighboring nodes, if there is no ambiguity.
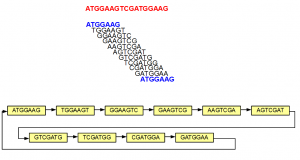
Two things can happen with removal of low-frequency k-mers by counting tools.
(i) Better quality assembly: If the low-frequency k-mer comes from noisy reads (for example, last nucleotide of a read being incorrect), those noisy regions go away from the graph and you get high-quality reconstruction. If, instead, you kept the low frequency k-mers, read extension would have stopped too soon and given very short contigs.
(ii) Introduction of errors: If, suppose, similar nucleotides are present at two places in the genome and the coverage is low in one of those two, removal of low-frequency k-mers will tell the assembler that there is no ambiguity. Then, the reconstruction of the contig will be erroneous.
I suspect (i) is far more dominant than (ii), but the answer always depends on quality of data, type of genome, size of genome, etc.