
Using de Bruijn Graphs for Short-read Assembly - (i)
Here is a part of the first chapter of the genome assembly book that I am working on. Please feel free to grab a copy from here, if you like. You can find more details at this link.
-———————————————————–
Chapter 1. Introduction
This book is written for thousands of biologists and bioinformaticians eager to gain new knowledge about the living systems using recent technological breakthroughs in nucleotide sequencing, known as next-generation sequencing (NGS). NGS technologies revolutionized biology by giving access to high- throughput sequencing instruments to most research institutions around the globe and even some well-funded individual labs. Such easy access to sequencing brought a dramatic shift in the way genetics research gets done. Merely a decade back, genome sequencing of eukaryotes used to be very long and expensive endeavor, for which hundreds of scientists had to raise funds together by writing joint white papers and petitioning to various government agencies. The tasks of sequencing and assembly were handled by dedicated sequencing centers, of which only a few existed around the globe. Naturally, the capacities at those sequencing centers were significantly constrained from high volume of requests. In contrast, today any scientist can get more nucleotides than the human genome sequenced within a week from a local facility for an insignificant price.
This rapid and somewhat unexpected democratization of sequencing capabilities left the biologists unprepared for the resulting data deluge. Every downstream analysis step, including sequence assembly, mapping and even storage of data, has now become a challenge. In previous arrangements, genome assemblies were performed by a few dedicated groups within the large sequencing centers, and those groups sheltered the biologists from various unpleasant aspects of handling large volumes of data. Many of those dedicated groups still remain the leaders in research on sequence assembly, but the scientists from other institutions are venturing into assembly and analysis to make sense of locally acquired short-read libraries. In fact, it is now nearly impossible to do high-quality genetics research in life sciences or medical sciences without proper knowledge of the bioinformatics tools. To make matters worse for the newcomers, the technological frontier in bioinformatics itself is moving fast with introduction of new tools almost every week. This book is written to make the lives of the life scientists easy by providing a simple introduction to the recent and ongoing developments of algorithms related to short read assembly.
Assembly procedure requires an understanding of sequencing technologies, algorithms and statistics
The genome is the primary informational unit of living organisms. It is packaged into multiple chromosomes. Those chromosomes get replicated and passed on from generation to generation to maintain continuity of the species. Natural selection works through random changes in the genome (mutation) and fixation of advantageous mutations within the population. Therefore, genome sequencing and assembly is an important step toward understanding the phenotypes and their modifications during evolution.
The genome assembly process, described in abstract terms, requires an understanding of three aspects - sequencing technology, algorithms and statistics. Let us see what roles they play.
Sequencing technologies
NGS technologies cover wide range of sequencing instruments, each offering unique benefits and challenges during assembly. The assembly process often requires combining sequences from multiple technologies. Moreover, many researchers are restricted to use certain technologies due to ease of access or cost. For these reasons, this book covers the differences between sequencing technologies and their impact on assembly process in chapter 4.
Algorithms
An algorithm consists of a set of rules to perform a given task. For example, young children learn algorithms to add numbers using carry and subtract using borrow. Conceptually, algorithms are not too different from the experimental protocols used by biologists. Their mastery requires two steps - understanding the concept and memorization through practice. Mechanization, such as using calculators to add and subtract numbers, cannot replace the value added from understanding of the concepts. Therefore, this book focuses on explaining the algorithms involved in assembly process. This point is further explained in the following section.
Statistics
Given that nucleotide sequencing is an experimental process, experimental variations and noise are always part of the data. Those artifacts manifest in the assembly procedure in two ways - (i) use of cutoff parameters to limit the amount of noise and (ii) use of statistical methods, such as averaging or hidden Markov model. Biologists are well familiar with those procedures from other contexts. This book explains the statistical aspects by first assuming error-free data and presenting the core assembly algorithm in the absence of noise (Chapter 3). Then the impact of noise and uneven coverage are explained in a later chapter (Chapter 5). By partitioning the assembly problem into three components, a biologist will be able to figure out whether his assembly quality can be improved by moving to different technology, better algorithm or higher coverage. Moreover, this book adds adequate amount of simulations and analysis of real data to allow readers play with various aspects and learn.
Why should biologists learn about assembly algorithms?
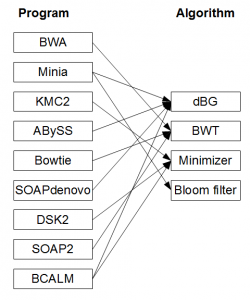
This books approach of focusing on algorithmic concepts departs from the current training practices in bioinformatics. In the current approach, the biologists analyzing NGS sequences typically learn to use and benchmark various software tools, such as Velvet, ABySS, Trinity, Oases, SOAPdenovo, Minia, Ray, IDBA, SPAdes, etc. to assemble the underlying genomes, transcriptomes or metagenomes. Even though all of those software programs use de Bruijn graphs to reconstruct genomes from NGS libraries, proper understanding of assembly algorithms is not needed to run
them. Is there any benefit for biologists in learning the inner workings of those assembly programs? The answer is an emphatic yes. Such an understanding helps in performing every step of the work including designing experiments, purchasing computing hardware, improving assembly quality, interpreting results and, most importantly, staying abreast of technological developments. Furthermore, NGS sequencing and assembly has become routine tools for analyzing transcriptomes (RNAseq) of non-model organisms, and the same benefits of learning about assembly algorithms apply there. The biggest advantage of understanding the algorithms is likely to come from the ability it provides in developing clever and unconventional experiments using NGS technologies. It appears that the full potential of NGS technologies is not being realized, because a large gap exits between those who think in terms of experiments and those who develop algorithms to analyze related large data. This book intends to close that gap and give an experimentalist power to think through all aspects of data analysis. Each of the above points is discussed in detail below.
Software tools change, but the algorithms remain more stable
Over the last three years, computational researchers developed many efficient programs to improve various assembly steps, but those programs remain under- utilized until they are fully incorporated into the existing pipelines. Switching from one program to another, or even deciding about proper benchmarking measures to test the effectiveness of new programs, is a major challenge for those trained with software programs. Proper knowledge of algorithms should make the task easier, because the underlying algorithms behind various programs stay more stable than the codes.
-———————-
-———————–
Knowledge of algorithms helps in experimental design
Designing of NGS experiments require making decisions regarding the mix of sequencing technologies, mix of mate pair libraries and read coverage. Such decisions are hard to make without a conceptual understanding of how the assembly gets done.
Understanding algorithms helps in purchasing adequate computing hardware
Bioinformaticians knowing the assembly algorithms can solve larger problems without buying expensive computer hardware. Exhausting available computer memory (RAM) is the first obstacle faced by new bioinformaticians working on de Bruijn graph-based assemblers. The most obvious and commonly sought solution of finding a computer with large RAM does not scale well with ever- increasing amount of data. A number of elegant algorithms were recently proposed by several bioinformaticians, but their implementation and incorporation into existing workflows require some knowledge of the underlying assembly process.
-———————–
-———————–
Improving assembly quality and interpreting results better
When a bioinformatician blindly runs a program to obtain results, it is unclear to him whether the gaps or errors in his assembly are due to shortcoming of technology, lack of coverage, execution of program or noise. Even with a familiar program, the quality of assembly can be greatly improved by searching over the entire parameter space, but some of those parameters can only be explained with reference to the de Bruijn graph structure.
RNAseq in non-model organisms
De Bruijn graph-based algorithms have become important for a new class of problems, namely transcriptome assembly (RNAseq). Transcriptomes did not need to get assembled in the older days of Sanger sequencing, because the sizes of ESTs were comparable to genes, whereas the gene expression was measured by microarray technology. High-throughput NGS can solve both problems together, but shorter NGS reads need to be assembled into genes, especially for organisms lacking underlying genomes. De Bruijn graph-based Trinity and Oases transcriptome assemblers are popular among researchers working on RNAseq data.
Using NGS technologies to solve novel problems
In 2010, a group from Fred Hutch Research center used NGS technologies to sequence the variable regions of human immune cells, namely T-cells and B-cells. Their method has been commercialized by Adaptive Biotechnologies, a Seattle based company. In 2012, a Stanford group came up with a method to sequence long reads synthetically using short read sequencing. Their technology was commercialized by Moleculo, a company that was later acquired by Illumina. Those are two of many examples of creative uses of NGS technologies. In all cases, the analysis of large amount of data was a critical component of development of the technology, and it often required designing new algorithms. It is expected that many other unconventional uses of NGS technology will be discovered in coming years, and an experimentalist with proper knowledge of algorithms will have an upper hand to think about creative applications.
-———————————————————–
Comments and suggestions appreciated. Also note that the sections posted here are draft versions to be incorporated in the next update of the book. I try to keep the number of updates to one per month or less.