
Using de Bruijn Graphs for Short-read Assembly - (ii)
Here is the next part of the first chapter of the genome assembly book that I am working on. Please feel free to grab a copy from here, if you like. You can find more details at this link.
-—————————————
Description of this book
Over the last four years, several introductory articles on de Bruijn graph- based assembly programs were published at homolog.us blog and they remain popular among our readers. For their convenience, the articles were compiled into tutorials and this book is an expansion of those tutorials to explore the same topics in further detail. This book describes how de Bruijn graph-based assembly programs work, explain why so much RAM is needed by the assemblers, show the impact of sequencing error on graph structures, discuss the differences between genome assemblers, transcriptome assemblers and metagenome assemblers and cover many recent algorithmic developments.
Chapters
i) Chapter The Genome Assembly Problem explains the concept of shotgun assembly and introduces the readers to various assembly algorithms, such as greedy strategy, overlap-layoutconsensus method and de Bruijn graph.
ii) Chapter De Bruijn Graph of a Genome explains how the de Bruijn graph of an already assembled genome looks like and demonstrates how perfect reads can be assembled using de Bruijn graph approach.
iii) Chapter Experimental Considerations covers sequencing technologies.
iv) Chapter Genome Assembly from Noisy Reads with Uneven Coverage explains genome assembly procedures for real-life reads containing errors.
v) Chapter Assembling Transcriptomes, Metagenomes and Highly Polymorphic Genomes shows how to solve other assembly problems.
vi) Chapter Faster, Better and Cheaper discusses various algorithmic improvements.
vii) Chapter In Depth Discussion of Three de Bruijn Graph-based Assemblers discusses the algorithms and codes of three assemblers in detail.
Algebraic notations are avoided as much as possible
To make this book more accessible to biologists, preference is given to pictorial style of narration over algebraic notations commonly used in bioinformatics papers. Readers should note that our chosen style is in no way inferior to algebra and can in fact provide better understanding of graphbased algorithms used for assembly. The preference for algebraic notations by computational papers is primarily to make the description more amenable to computer programming.
Living electronic book
This book chooses a publishing style that allows regular updates of the book to keep content up to date with the rapidly advancing NGS bioinformatics. Unlike other technical books, whose contents are static once the book is published, this electronic book will continue to get updated with new information, and readers will be able to download and read the latest version at no extra cost. Leanpub, as an online publisher, allows readers to take advantage of this feature. In addition, the book provides other advantages of electronic publishing, such as using hyperlinks to go between chapters, being able to use search capabilities of electronic reader, etc.
Core concepts are reinforced through repetition
The primary aim of this book is to make sure the readers fully understand and remember the core concepts after reading this book. Therefore, entire chapters in the early part of the book are dedicated to explaining ideas covered in at most one or two sentences in technical papers, whereas a later chapter Faster, Better and Cheaper condenses discoveries presented in many advanced papers. If the readers grasp the core concepts explained in earlier chapters, they will be able to go through the advanced papers themselves with some contextual help from our explanations. Explanations for some of the key concepts are repeated in multiple places to facilitate understanding, and the codes and exercises are expected to further reinforce the learning.
Regarding the exercises
Code - Pandoras Toolbox for Bioinformatics
Many useful bioinformatics programs are scattered all over the internet and it is not easy for a newcomer to locate them without some guidance. Therefore, Pandoras Toolbox for Bioinformatics brings a number of useful codes together. The toolbox is named after Symbion pandora, an unusual animal discovered in 1995.
-—————————-
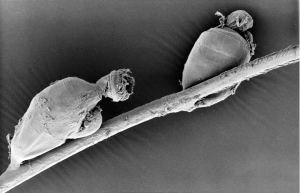
Fig 3. Symbion pandora (image courtesy Dr. Peter Funch)
-—————————-
Within Pandoras Toolbox, three programs - kmc, bcalm and tip-cutter - will be frequently used in this book. Graphviz, a fourth program, is useful for viewing the de Bruijn graphs.
Program
Use
kmc
k-mer counting
bcalm
de Bruijn graph compaction
tip-cutter
removing tips from the graph
bandage
viewing de Bruijn graph
bwa
aligner
samtools
for processing alignments
The program kmc takes one or more read libraries as input and counts the number of k-mers of certain size (k) within them. The following step produces two binary files - kmers.kmc_pre and kmers.kmc_suf. K-mers present only once in the libraries are removed.
`1 % mkdir tmp
2 % kmc -k25 [library] kmers tmp
`
The program kmc_dump_bcalm extracts k-mers present 4 or more times from the binary output of kmc and saves in text file in.dot in format suitable for bcalm.
1 % kmc_dump_bcalm -ci4 kmers in.dot
The program bcalm builds a de Bruijn graph from the k-mers in in.dot and compresses their linear portions to generate out.dot. Then tip-cutter removes all tips from out.dot and saves in out-clean.dot.
`1 % bcalm in.dot out.dot
2 % tip-cutter out.dot > out-clean.dot`
Data - E. coli and Electrophorus
The following data sources will be used in the examples given in the book.
i) Hydrogen atom
This small 1,000nt E. coli sequence along with associated reads has been provided by the authors of SPAdes as a toy set, and it is being used here for demonstration purposes. The genome and reads can be downloaded from here.
ii) E. coli genome and reads (SPAdes)
-—————————-

Fig 4. A segment of E. coli genome. Blue in the figure shows repetitive regions. Codes for constructing similar plots for the other parts the genome are available from homolog.us website.
-—————————-
The fully assembled E. coli genome is only 5 megabases is size and can be used to learn about algorithms. A shor read dataset covering the E. coli genome at 1000x can can be downloaded from the SPAdes website?.
iii) Electric eel (Electrophorus electricus) reads (SRA)
Electrophorus genome is about 700 megabases long. The short read sequences can be downloaded from the NCBI SRA website?.
Other online resources for learning
Apart from the books within our introductory series, a number of online resources exist for those interested in learning bioinformatics on their own. They are classified below as introductory resources, intermediate resources and advanced resources.
Introductory resources
Rosalind and Coursera
-—————————-
![]()
Rosalind
-—————————-
Rosalind?, developed by a team led by Prof. Pavel Pevzner, allows students to learn bioinformatics
online through problem solving. Professor Pevzner and his student Phillip Compeau also developed
an online bioinformatics course through Coursera that is freely available from youtube?.
Software Carpentry
Many newcomers to bioinformatics do not have any background in running computer programs,
and especially those programs requiring experience with command line and unix operating system
pose particular problems. Software Carpentry created a number of useful tutorials? to simplify the
learning process.
R and Ipython notebook
R? is an useful computational tool, where statistical analysis and plotting of data can be easily done without knowledge of programming.
Ipython notebook? is a web-based interactive computational environment to help with executing codes, adding plots and writing text and equations.
Seqanswers
In online forum Seqanswers, users ask question about various bioinformatics programs or analysis
approaches and get answers from the experts.
Biostar
Biostar is an online forum like seqanswers, but it follows a different format. Each answer gets ranked by the members of the community so that the correct or most useful answer rises to the top. It creates an useful knowledge-base for future reference.
Intermediate Resources
The following blogs provide useful information on bioinformatics algorithms.
Homolog.us
Homolog.us blog presents useful information about the latest research in bioinformtics.
Heng Li
Dr. Heng Li of Broad Institute maintains two informative blogs - one at github? to cover bioinformatics algorithms and one at Attractive Chaos? to cover his lightweight klib library and other computer science-related topics.
Dazzler blog
The Dazzler blog? maintained by renowned researcher Gene Myers releases information about his long-read assembly tools.
C. Titus Brown
The blog maintained by Dr. C. Titus Brown? discusses benchmarks and analysis of various useful bioinformatics programs.
Advanced resources
Arxiv and biorxiv
The preprint websites arxiv? and biorxiv? have become invaluable resources for those doing advanced research in bioinformatics. These days, most computational researchers submit their papers at one of those repositories long before they get formally accepted at journals.
Github
Many bioinformaticians doing cutting-edge research do not write blog posts to describe their work, but often maintain very active github sites to publicly post their codes. Others may look into the codes to learn about the problem- solving approaches used by them. The links to a number of useful github pages are provided below.
Bioinformaticians and their github pages
GATB?
Rayan Chikhi
Heng Li
Jared Simpson
Gene Myers?
Alex Bowe?
Jason Chin?
Dinghua Li?
Acknowledgements for this book
The author is thankful to Rayan Chikhi, Jason Chin, Kevin Karplus, Anton Korobeynikov, Heng Li, Ruibang Luo and Jared Simpson for helpful discussions over the years or comments on this manuscript. The author also thanks the readers of the homolog.us blog and Twitter followers for bringing informative links on latest research to his attention.
Further readings
-—————————————-
http://homolog.us
https://github.com/homologus/Pandoras-Toolbox-for-bioinformatics
https://github.com/homologus/Hydrogen-atom
?http://spades.bioinf.spbau.ru/spades_test_datasets/ecoli_mc/
?http://www.ncbi.nlm.nih.gov/sra/SRX554971
?http://rosalind.info/problems/locations
?https://www.youtube.com/user/bioinfalgorithms
?http://software-carpentry.org
?http://www.r-project.org
?http://ipython.org/notebook.html
http://seqanswers.com
http://biostars.org
http://homolog.us/blogs
?http://lh3.github.io/
?https://attractivechaos.wordpress.com/
?https://dazzlerblog.wordpress.com/
?http://ivory.idyll.org/blog/
?http://arxiv.org
?http://biorxiv.org
?https://github.com/GATB/
https://github.com/rchikhi
https://github.com/lh3
https://github.com/jts/
?https://github.com/thegenemyers/
?https://github.com/alexbowe/
?https://github.com/cschin/
?https://github.com/voutcn