
De Novo Hybrid Assembly of PacBio and Illumina Reads - (i)
We are currently working on a project to assemble a large genome (~2GB) de novo from Illumina and PacBio reads. We decided to post our notes here for the benefit of others.
Our collaborators sequenced huge amount of Illumina reads (~200x coverage, 100nt X 2 paired-end and mate-pair) and about 7-8x coverage of PacBio (average length 3KB) with plan to sequence more. Based on their earlier experience, the genome has large amount of repeats and therefore they expect hybrid assembly to be beneficial.
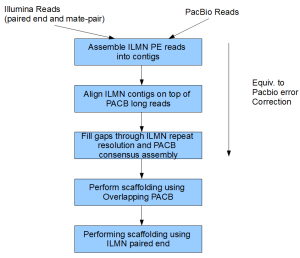
Our current work-flow for assembly is shown below. Some of the steps are in the form of private scripts. We are also developing an automated software package in parallel to incorporate those steps and will release them as soon as they are ready for public consumption.
-——————————————————
Step 0. Moving around the Read Libraries
There was an important ‘Step 0’ before we could start. Since the ILMN data files are so huge, downloading/uploading them took enormous time and the fastest way turned out to be shipping hard-drives in car/truck/plane from place to place. So much for living in internet age !!
-——————————————————
Step 1. Assemble ILMN Paired End Reads into Contigs
Our collaborators previously used ALLPATHS to assemble ILMN paired end and mate pair reads into both contigs and scaffolds. The assembly step took ~500GB RAM !!
We decided to redo this step using Minia for four reasons -
(i) Minia is memory-efficient and our server has only 10GB of RAM.
(ii) We understand Minia code and algorithm well. After some bad experience with Velvet scaffolder in the past, we decided not to use any assembler, whose code we did not understand.
(iii) Minia can use very long k-mers to assemble reads and that is better for repeat resolution. ALLPATHS claims to be k-mer independent, but it appears to use 25-mer as its default k-mer size.
(iv) We expect that understanding the code will be very helpful in later steps, where we try to fill gaps based on PacBio/Illumina combined assembly.
The Minia step took about three days (half of which were spent in k-mer counting) and produced a contig assembly with total size of ~710Mb (N50=~1Kb).
-——————————————————
Step 2. Align ILMN Contigs on top of PACB Long Reads
After completing Minia assembly, we aligned the contigs on top of PACB long reads. We use our unpublished algorithm (takes minutes), but you are free to use BLASR provided by PacBio. Mark Chaisson, who developed BLASR, is currently working on an ultrafast version that may have already been incorporated into the latest github release.
-——————————————————
We will continue this discussion in future commentaries, where we will discuss the patterns of errors seen in ILMN and PACB reads and assemblies.