
Hybrid Assembly - (ii): The Error Models of PacBio Reads
Now that we have contigs assembled from short Illumina reads aligned on to long PacBio reads, the question of which one to trust often pops us in our mind. Let us explain the issues more clearly.
-——————————————————————-
A. Often we come across regions, where 3/4th of a long Illumina contig matches very well with the PacBio read (after allowing for 85% of error rate), but the remaining contig is not seen anywhere nearby.
Possibilities:
Illumina is correct.
One can make a case that Illumina contig is built from hundreds of short overlapping regions, and therefore the Illumina contig is more accurate.
PacBio is correct.
One can also argue that the particular genomic region is different in two chromosomes and the PacBio read is capturing a different chromosome compared to what is assembled from Illumina. Possibly the chromosomal region has a large insertion/deletion.
-——————————————————————-
B. We also come across regions, where the Illumina contig matches PacBio closely, but has a large gap inside. The gap is usually filled with homopolymers.
Possibilities:
Once again, one can argue about both possibilities mentioned in A.
-——————————————————————-
C. Third case of ambiguity is multiple copies of the same Illumina contigs matching a PacBio contig.
Possibilities:
PacBio is correct.
By its design, k-mer based de Bruijn graph assembly compresses duplicated regions into one block. Therefore, the contig assembly method used for Illumina reads is incapable of resolving tandem repeat regions.
Illumina is correct.
PacBio technology circularizes the chromosomal fragments and then goes over them again and again. Therefore, the raw PacBio reads have multiple copies of the same chromosomal region, but the initial processing step splits them into different reads. It is possible that the processing step may have missed a few circularized junctions.
-—————————————————————————-
Yesterday, we took time to meticulously work through a few cases to understand what is going on, and we will share the examples here. They are anecdotal cases rather than systematic analysis of the entire data set, but will illustrate the points mentioned in A, B and C to help you appreciate the issues.
Error Case B.
Here are two examples of large homopolymer difference between long PacBio reads and assembled Illumina contigs. In both alignments, the sequence with ‘len’ in the name is the assembled contig and the other sequence is the PacBio read.


Notes.
1. In both cases, PacBio had large insertions and not deletion compared to Illumina contig.
2. We manually checked about 15 alignments and found only two examples of such homopolymer expansion.
3. The genome is highly repetitive, and therefore such long stretches of Ts and Cs are not unexpected.
4. We BLASTed both the Illumina contig and the PacBio read against all other PacBio reads. We did not see any single match of the PacBio region with another different read, whereas we found several hits with the assembled Illumina contig. That makes us believe that assembly from Illumina is correct and PacBio is wrong here. We are not seeing an effect differences between two copies of the chromosomes.
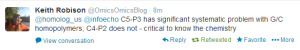
We checked with Jason Chin, whether he sees similar differences while doing genome assembly.
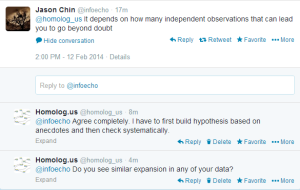

Edit. Here is the beauty of twitter. We can use our collective brains to solve problems.
-—————————————————————
Error Case A.
The contig ‘7117__len__1384’ assembled from Illumina is 1384 nt long and it matches nine PacBio reads longer than 3KB (IDs: 10, 11, 112003, 491553, 655988, 772698, 1069707, 1744092, 2033153). We tried to align those PacBio reads on top of each other to build consensus and observed something puzzling.
1. Three PacBio reads ( 772698, 11(R), 491553(R) ) aligned well with each other over their entire length. How about the rest?
2. To find out what is going on with the remaining five reads, we aligned each of the individually with the longest PacBio read (772698) from the above group of four.
i) Half of read #10 aligned well with #772698 and the remaining half did not. You can see the ‘good alignment-bad alignment’ boundary in the following image.

What does it mean? Possibly we have a chimeric PacBio read or heterozygosity or something else altogether. We took apart the part of read #10 (~2500 nt) that did not align well and compared against all PacBio reads and all assembled Illumina contigs using BLAST (blastn). This comparison did not give a single hit, which suggests that the 2500 nt non-matching half is not part of the genome. Clearly, this was an example of sequence error.
ii) Two other PacBio reads - #1069707 and #655988 showed similar pattern of partial matching with #772698, and we initially assumed that they were also bad sequences. Interestingly, those two reads aligned well with each other. So, you see a nice clustering effect of three reads (772698, 11(R), 491553(R)) aligning with each other and two others (1069707 and 655988) aligning with each other, but the entire group have only 50% overlap.
We also found that the non-matching halves of #1069707 and #655988 aligned well with an Illumina-assembled contig - 122456__len__2371. Therefore, unlike #10, these two are legitimate reads and represent some kind of complex structure within the genome.
iii) The remaining two PacBio reads aligned well with #772698 on their two edges, but had large gaps or mismatches in the middle. An example is presented below.

Does the above case represent sequencing error, or genomic complexity? We are yet to figure out.
As you can see, a good hybrid genome assembler needs to resolve many complex situations like above to properly assemble the genome. We did not pick 7117__len__1384 through any systematic process to demonstrate assembly complexity. It was rather the first contig we randomly selected to investigate in detail and it may represent average case of complex problem in our hand.
Many large eukaryotic genomes were assembled from Illumina-only reads and it is unlikely that they had complex situations like above properly resolved. Will it be necessary to revisit all those genome projects or is the assembly we are currently involved in unusually complex?