E. coli Nanopore Data Release and #baltiandbioinformatics Nanopore Conference
Nick Loman uploaded his E. coli nanopore data on GigaDB.
Here we present a read dataset from whole-genome shotgun sequencing of the model organism Escherichia coli K-12 substr. MG1655 generated on a MinION device with R7 chemistry during the early-access MinION Access Program (MAP).
Three sequencing runs of the MinION were performed using R7 chemistry. The first run produced 43,656 forward reads (272Mb), 23,338 (125Mb) reverse reads, 20,087 two-direction (2D) reads (131Mb), of which 8% (10Mb) were full 2D. Full 2D reads means that the complementary strand was successfully slowed through the pore.
The R7 protocol has a modification to increase the relative number of full 2D reads (NONI). To exploit this, two new libraries were produced which included an overnight incubation stage (ONI-1 and ONI-2). Each library was run on an individual flow cell. This resulted in 6,534 & 8,260 forward reads, 2,171 & 2,945 reads and 1,740 & 2,394 2D reads (27%, 29%). In this case, 50% and 41.8% of reads were full 2D respectively. The mean fragment lengths for 2D reads from the three libraries were 6,543 (NONI) 6,907 (ONI-1) and 6,434 (ONI-2).
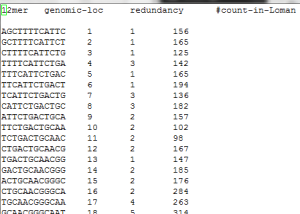
We have taken the reads, created a kmer distribution and then merged that distribution with the kmers from E. coli reference genome (MG1655-K12.fasta). The kmer distribution file for reads can be downloaded from here and genome map can be downloaded from here. A snapshot is shown below:
(column 1: 12mer, column 2: genomic loc, column 3: redudancy in genome, column 4: kmer count in Nick Loman’s data)
Those two files should tell you a lot more about the technology and its potential than many talks, papers and blog posts. So, readers are encouraged to take a look.
Few observations:
1. Good match with genome in most locations
Based on kmer distribution of reads, we checked for kmer coverage of genome and found kmers present from almost all locations of the genome.
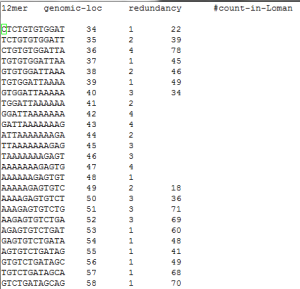
2. Poly-A regions are missed.
If point 1 gives you any confidence in the data (or our analysis), you are fooled. Please note that given the number of reads and short length of 12mers, completely noisy data are also expected to have almost all kmers from the genome. However, complete lack of k-mers from some parts of genome is informative and can be definitely ascribed to base-calling errors. We found a few such missing regions, and almost always they had poly-A stretches.
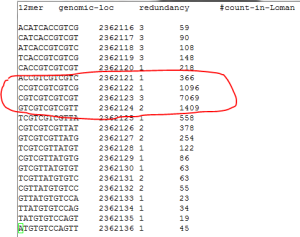
Here is the opposite extreme - genomic regions with too many unexpected kmers in the reads.
3. Kmer distribution - read vs ref
If almost every kmer is expected to appear among the reads, how can we tell whether they contain any information? The following figure could be of help. Dotted line shows occurrence frequency of all kmers from the reads, and solid line shows the same for only those kmers that actually appear in the genome. The solid peak is shifted to the right and that means the kmers in the genome appear more frequently in the reads than random kmers.
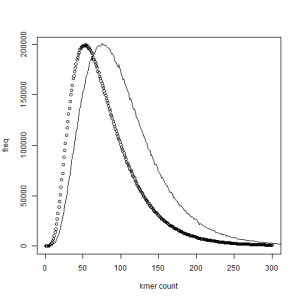
4. Unusual kmers in the reads, not from the genome
The reads also have some kmers appearing at high frequency, which are not at all present in the genome.

Analysis 1-4 should be helpful in choosing an appropriate aligner rather than shooting blindly. We will add more, as we analyze data in more detail.
-————————————–
On #baltiandbioinformatics Nanopore Conference
1. Very impressive. This is the future !!
‘This’ refers to Google hangout way of running conferences, which anyone can attend from his/her bedroom and Bill Clinton with his cigar :)
Scott Edmunds’s GigaDB, which we covered earlier, turns out to be another tech winner in the publishing world.
2. Nanopore Technology
Two highlights -
a) SPAdes assembled E. coli using nanopore+ILMN and got good results. Note - they needed ILMN and so did Schatz.
b) Error rate figure -

Clive Brown’s talk is very impressive and he is beaming with infectious optimism. Hopefully Loman’s tests will be able to test how fast that infection spreads :).
His vision is right, but bioinformatics remains the weak point in this project as explained next.
3. Cost of sequencing
If each nanopore stick costs $1000, cost of sequencing is high compared to Pacbio (and Illumina). The mobility nanopore provides is truly revolutionary, but for his vision to work, the price point for the sticks has to be way lower.
We have no idea what the manufacturing costs for those sticks are, but let us assume that he can provide them for $10 a pop. In that case, to maintain a cash flow of $100M or higher (to justify the valuation), data management and analysis has to be the strong point of the company. So, essentially ONT becomes a bioinformatics company.
-——————————–
Edit.
Interesting viewpoint of the CEO of company that wants to pass itself as a ‘small startup’ from a garage.