Analysis of NGS miRNA Data – a Paired-ended Library
Today, we will analyze a paired-end miRNA library sequenced using Illumina technology. Paired-ended libraries give twice as much sequences as single- ended, but their strand-orientations need to be carefully considered for analysis. In an earlier commentary, we discussed the strand-orientation issues for Illumina paired-ended reads from genomic regions, and showed that two reads of a pair aligned with opposite strands of the genome. Also, the reads were typically inward-looking for paired end reads, but outward-looking for mate pairs.

Those conventions vary for other NGS technologies, such as ABI SOLiD, 454, and need to be kept in mind before using any genome assembly package. For example, the ‘reverse_seq’ parameter is used for specifying the ‘inward-looking’ and ‘outward- looking’ orientations in SOAPdenovo2.
MiRNAs are too tiny even for NGS short read sizes. Therefore, their lengths are increased by adding linkers before sequencing is performed. Earlier (here and here), we discussed how variable linkers are added to sequence multiple samples together, and explained the process with an example of a multiplexed 7-tissue single-ended library.
What does a paired-ended Illumina library of miRNA sequences represent? Instead of giving you the answer, we will figure out the protocol from real data using the procedure presented in our earlier commentary.
We have an Illumina library consisting of 101,853,001 x 2 reads from miRNAs of a vertebrate organism. Each read is 101 nt long.
Linker sequences for set1 and set2
The analysis is simple, because all one needs to do is take a small subset of reads and find any common short k-mer (k is anywhere from 25 to above). We did the analysis on the first set and found the common linker sequence - ‘TGGAATTCTCGGGTGCCAAGGAACTCCAGTCACCAGATCATCTCGTATGCCGTCTTCTGCTTGAAAAAAA’ in about half of the reads. The sequence is same as the linker used for single- ended reads discussed in our previous commentary, and it was present near 3’ ends of all reads.
For the second set of paired-end library, we found a common sequence - ‘GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATTAAAAAAA’ Two things to note here -
(i) Here also the linker was present near 3’ ends of all reads,
(ii) Linker for second set was 8 nucleotides shorter than the linker for the first set. It is possibly due to its not containing the variable bases in the middle used for multiplexing.
Finding reads containing the linker sequences
We separately searched through all reads from first and second libraries to find matches with the respective linkers. Up to two mismatches were allowed. A total of 49,761,000 or 49% of the reads had matches with the full linker in the first library. The corresponding number for the second library was 46,340,069 (45% of all reads).
If you remember our previous commentary on single-ended reads, it had 39% of all reads matching the full linker. Do we have an improvement in the current experiment? Not really, as will be clear from the following chart.
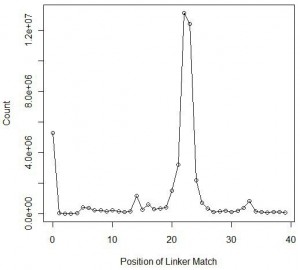
Here we are plotting the positions within the reads of second set, where the linker matched. As expected, the chart shows a large peak at nucleotide 22-23, the average size of a mature miRNA. However, unlike the previous experiment, a large peak appeared near nucleotide=0. In those cases, no miRNA attached to the ends of the linkers, and instead the instrument sequenced the linkers only.
Just like our prior analysis on single-ended reads, we also determined how many reads from the first set matches a truncated version of the linker (‘TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC’). The number improved to 65,452,376 or 64% all reads. Like before, much of the added contribution came from short RNA sizes of 60-65 (see following figure).
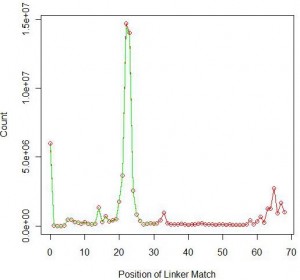
In the above figure, green shows the matching position of the full linker and red shows the matching position of truncated linker.
Comparison of reads containing full linker from set1 and set2
We have seen that 49,761,000 or 49% of the reads from set1 of paired-ended library had the the full linker. For the second library, 46,340,069 (45% of all reads) had its corresponding linker. Given that each read from set1 has a sister read in set2, one should ask how those 49,761,000 and 46,340,069 are related. One extreme possibility (disjoint) is that those reads with linker match in set1 have sister read in set2 with no linker match. At the other end (fully overlapping), it is possible that the groups 49,761,000 and 46,340,069 are highly overlapping. Only those reads that had linker match in set1 have linker match in set2. Also, there are many other possibilities in between suggesting that two groups are unrelated.
Based on analyzing our data, we find that our reads are very close to fully overlapping end. Over 83% of reads with linker1 match also had linker2 match.
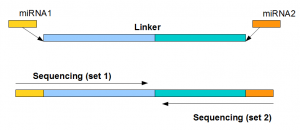
miRNAs from the first and second set
Finally, we look at the miRNA sequences from the first and second set. They are found to have the pattern shown in following figure. The miRNAs from set1 usually had the same strands as the miRNAs in miRBase repository, whereas miRNAs from set2 were reverse stranded and the linker was added to the 3’ end of the reverse stranded sequence. Also, we compared the expression patterns of miRNAs derived from set1 and set2 and found them to be identical.

What is going on?
For paired ended sequencing, one long linker (containing both linker sequences shown above near two ends) is added to the sample containing miRNAs. MiRNAs ligate to both ends of the linker, and then the sequencing instrument decodes the combined sequence from its two ends. The sequencing done from the 5’ end shows the miRNA in proper strand, whereas the sequencing done from 3’ end is done from reverse strand and it inverts the miRNA sequence.