
Standard Steps for RNAseq Experiment
A collaborator asked us questions about various steps of a transcriptome (RNAseq) experiment. For everyone’s benefit, here we are presenting the complete RNAseq pipeline with short comment on each step. Typical bioinformatics program used for each step is also mentioned. If you use something different, please feel free to mention in the comment section.
For illustration, we will use one of our array experiments published several years back. In the experiment, we used custom-designed arrays to understand regeneration of Chlamy flagella.
Green alga Chlamydomonas loses its flagella under stress. Then, it regrows the flagella over one hour. Which genes are active in regrowth of flagella? The question is highly important for human health, because the chlamy flagella have the same set of genes as cilia in human body and the genes are highly conserved.
Let us say, we have three different Chlamydomonas-like species, where we like to replicate the above experiment. None of those species have their genomes sequenced and that makes RNAseq the perfect tool for exploration.
We also assume that for each species, we will study four different time points
- (i) Right after losing flagella and at the onset of regrowth, (ii) 15 minutes after beginning of regrowth, (iii) 30 minutes after beginning of regrowth, (iv) 1 hour after beginning of regrowth, when full flagella formed.
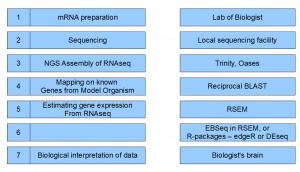
Going back to our flowchart,
Step 1. We will need RNA from each of time point and each Chlamy-species. In total, 4x3=12 samples need to be prepared. We are leaving out the question of biological replicates, etc.
Step 2. All 12 RNAseq samples need to be sent to local sequencing facility to be sequenced. Let us say, we use Illumina GA2 technology. At the completion of step 2, sequencing center gives back 12 fastq files full of [ATGC]s.
Step 3 The advantage of RNAseq experiment (over microarray) is that it allows one to find the gene sequences in a new organism, as well as give the expression profile. Determining gene sequence through assembly of short reads come first.
Trinity has become an effective assembly tool for transcriptome reads from unknown organisms. RAM-permitting, we will pool together all reads for each specie from four different time points to do the gene assembly. Afterward the reads from individual time points can be mapped on to assembled genes for time-course estimation.
Step 4 At this point, we have a set of assembled genes from Trinity, but we do not know anything else about them. The best strategy is to run reciprocal BLAST on Chlamydomonas reinhardtii, because its genes are well-annotated.
Step 5 and 6 Now that we have a reduced set of genes, we can go back to our original question of finding differentially expressed genes. Firstly, we need to map the raw reads back to assembled genes. Then, we need to identify genes that are significantly up or downregulated in each sample.
One approach is to use Bowtie to map reads on to the assembled genes, and then use R packages like EBSeq, edgeR or DESeq to do the statistical analysis. The alternative is to use RSEM package.
RSEM uses EBSeq only, and with the following justification -
Popular differential expression (DE) analysis tools such as edgeR and DESeq do not take variance due to read mapping uncertainty into consideration. Because read mapping ambiguity is prevalent among isoforms and de novo assembled transcripts, these tools are not ideal for DE detection in such conditions.
EBSeq, an empirical Bayesian DE analysis tool developed in UW-Madison, can take variance due to read mapping ambiguity into consideration by grouping isoforms with parent genes number of isoforms. In addition, it is more robust to outliers. RSEM includes the newest version of EBSeq in the folder named EBSeq.
Step 8 Finally, we have a short table with -
(a) IDs and expressions of genes differentially expressed during the time- course study,
(b) functional annotation of those genes,
(c) sequences of the genes.
Time to open the Champagne bottle :)