
Genomic Analysis with Spark - A Few Examples
Yesterday’s post on Spark mentioned that the technology is not being used in bioinformatics. That is not entirely correct, and we came across a (small) number of mentions here and there.
Many time-consuming analyses of next generation sequencing data can be addressed with modern cloud computing. The Apache Hadoop-based solutions have become popular in genomics due to their scalability in a cloud infrastructure. So far, most of these tools have been used for batch data processing rather than interactive data querying.
The SparkSeq software has been created to take advantage of a new MapReduce framework, Apache Spark, for next generation sequencing data. SparkSeq is a general-purpose, flexible and easily extendable library for genomic cloud computing. It can be used to build genomic analysis pipelines in Scala and run them in an interactive way. SparkSeq opens up the possibility of customised ad hoc secondary analyses and iterative machine learning algorithms. This paper demonstrates its scalability and overall very fast performance by running the analyses of sequencing datasets. Tests of SparkSeq also prove that the use of cache and HDFS block size can to be tuned for the optimal performance on multiple worker nodes.
Availability and Implementation: Available under open source Apache 2.0 license: https://bitbucket.org/mwiewiorka/sparkseq/
-———————————————-
2. Genomic Analysis Using ADAM, Spark and Deep Learning
-———————————————-
Readers may also take a look at Heng Li’s answer to the following question at biostars -
Why is Hadoop not used a lot in bio- informatics?
Heng Li:
Most of applications you mentioned can be and have already been implemented on top of hapdoop. A good examples is the ADAM format, a hapdoop friendly replacement of BAM, and its associated tools. They are under active development by professional programmers. Nonetheless, I see a few obstacles to its wider adoption:
1. It is harder to find a local hadoop cluster. My impression is that hadoop really shines in large scale cloud computing where we have a huge (virtual) pool of resources and can respond users on demand. In a multi-user environment given limited resources, I don’t know if a local hadoop is as good as LSF/SGE in terms of fairly balancing resources across users.
2. We can use AWS, google cloud, etc, but we have to pay. Some research labs may find this unfamiliar. Those who have free access to institution wide resources would be even more reluctant.
3. Some pipelines are able to call variants from 1 billion raw reads in 24 hours with multiple CPU cores. This is already good enough in comparison to the time and cost spent on sequencing. There is not a huge need of better technologies. In addition, although hadoop frequently saves wall-clock time due to its scalability, at times it wastes CPU cycles on its extra layer. In a production setting, the total CPU time across many jobs matters more than the wall-clock time of a single job. Some argue that the compute-close-to-data model of hadoop is better, but for many analyses we only read through data once. The data transferred over network is the same as dispatching data in the hadoop model.
4. Improvements to algorithms frequently have much bigger impact on data processing than using a better technology. For example, there is a hadoop version of MarkDuplicates that takes much less wall-clock time (more CPU time, though) than Picard. However, recent streamed algorithms, such as SamBlaster and the new Picard, can do this faster in terms of both CPU and wall-clock time. For another example, there is a concern with the technical difficulty in multi-sample variant calling, so someone developed a hadoop-based caller. When it comes out, GATK has moved to gVCF, which solves the problem in a much better way, at least for deep sequencing. Personally, I would more like to improve algorithms than to adapt my working tools to hadoop.
For some large on-demand services, hadoop from massive cloud computing providers is hugely advantageous over the traditional computing model. Hadoop may also do a better job for certain bioinfo tasks (gVCF merging and de novo assembly coming into my mind). However, for the majority of analyses, hadoop only adds complexity and may even hurt performance.
Heng Li works for Broad Institute, which processes considerable amount of data
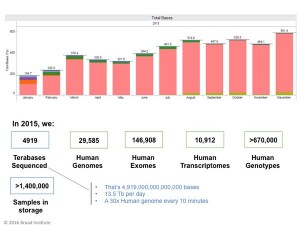
Given that his answer is 15 months old, maybe Broad is evaluating the technologies now. The real challenge is the lack of competent people with knowledge of both bioinformatics and fault-tolerant hardware technologies. Hopefully, our posts will generate some awareness about the usefulness of these new approaches.