Note: These tutorials are incomplete. More complete versions are being made available for our members. Sign up for free.

The World of Mapping Programs
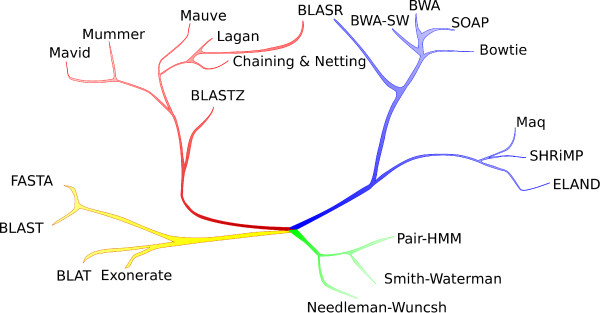
In an earlier section, we covered the above chart from <A href=http://www.biomedcentral.com/content/pdf/1471-2105-13-238.pdf>Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory</A> by Mark J Chaisson and Glenn Tesler. Now that we are familiar with the algorithms, we can go over various branches in the order of their historical developments and learn about how the developments in computer science impacted them.
Green branch was the earliest to develop. It includes dynamic programming algorithms such as Needleman-Wunsch and Smith-Waterman. Those algorithms gave exact solutions for global and local alignment, but ran too slow for all practical purposes.
The yellow branch developed over the following twenty years to allow biologists to solve real-world problems. Among those, BLAT and Exonerate dealt with alignments, where query and reference matched almost exactly, whereas BLAST and FASTA handled some amount of mismatch.
The red branch developed around the turn of the century, when the computer science developments involving suffix arrays became efficient. SSAHA, a precursor to Maq, SHRiMP and ELAND was written at the same time and it leveraged availability of large amount of RAM to hold the index in hashed form. SHRiMP, MAQ and ELAND were similar algorithms applicable for large volume of next-generation sequencing data.
The blue branch with BWA-SW, SOAP, Bowtie, etc. used Burrows Wheeler transform, and its use followed the discovery of FM indexing method by computer scientists. BLASR also used Burrows Wheeler transform, but was custom-designed for reads from PacBio technology.
We will go over each program and discuss their implementations.