Note: These tutorials are incomplete. More complete versions are being made available for our members. Sign up for free.

De Bruijn Graph of a Small Sequence
De Bruijn graph is an efficient way to represent a sequence in terms of its k-mer components. In an assembly problem, a de Bruijn graph is constructed from an NGS library, and then the genome is derived from the de Bruijn graph. To ease our understanding, in this section, we will check what the de Bruijn graph of an already assembled genome looks like. How short reads fit into the picture will be covered in Section 3.
A de Bruijn graph can be constructed for any sequence, short or long. The first step is to choose a k-mer size, and split the original sequence into its k-mer components. Then a directed graph is constructed by connecting pairs of k-mers with overlaps between the first k-1 nucleotides and the last k-1 nucleotides. The direction of arrow goes from the k-mer, whose last k-1 nucleotides are overlapping, to the k-mer, whose first k-1 nucleotides are overlapping.
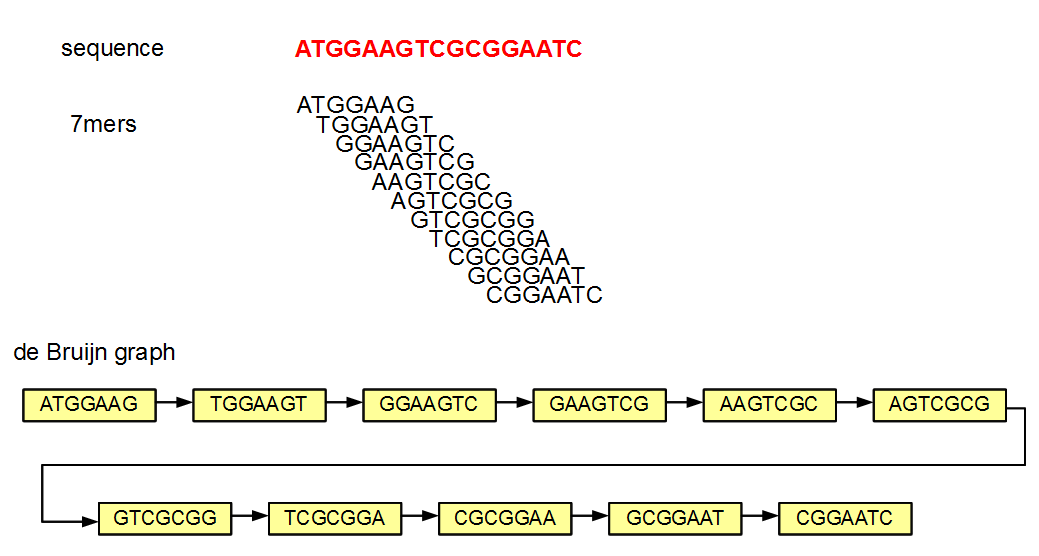
The method is best explained by an example. For sake of simplicity, we ignore the issue of double-strandedness of nucleotides here. In the above figure, we constructed the de Bruijn graph of ATGGAAGTCGCGGAATC by splitting it into all k-mers (k=7) and then by constructing a directed graph with those 7-mers as nodes. Edges are drawn between node pairs in such a way that the connected nodes have overlaps of 6 (=k-1) nucleotides. In our example, only the adjacent 7-mers from the original sequence got connected in the graph.
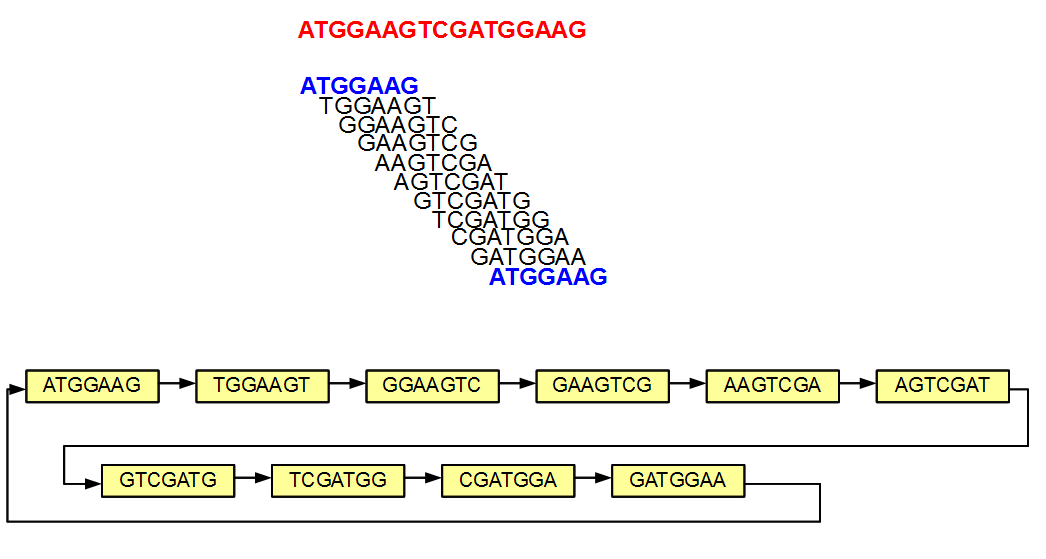
The above example is simple, because none of the 7-mers appeared more than once in the original sequence. In the next example, the 5′-most and 3’-most 7-mers are identical (both marked in blue). The de Bruijn graph has one less node due to merger of those two identical nodes. Like before, the adjacent nodes in the original sequence are still connected in the graph, but in addition the graph forms a loop by connecting its two ends.
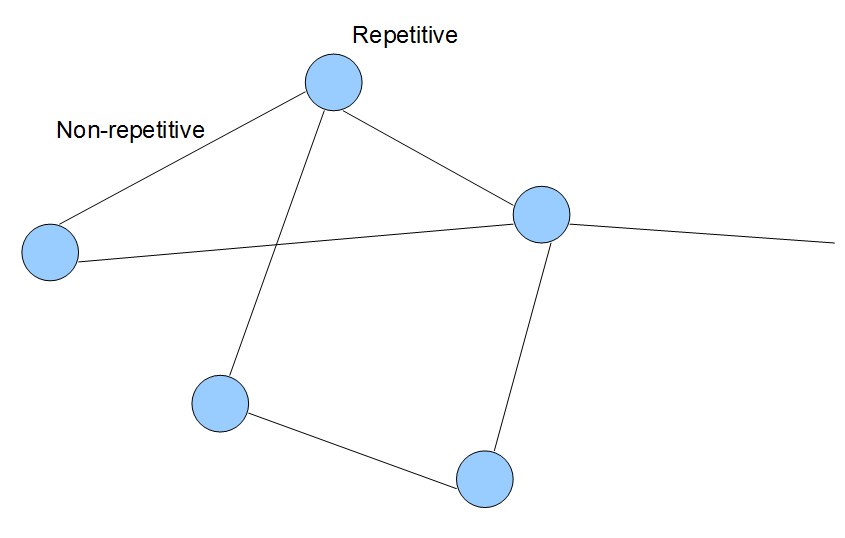
The steps shown above can be repeated to construct de Bruijn graph of any large genome with k-mer of any size. First, the genome is split into its k-mer components. Then various k-mers are connected based on whether they have k-1 common nucleotides. For prudently chosen k-mer size, topology of de Bruijn graph of a large genome will not look too different from the above figures. Vast majority of links will connect only the adjacent k-mers from the genome, whereas distant parts of the genome will get connected mostly in repetitive regions. The general topological structure is shown in the following figure, where each line and circle represents collection of many k-mer nodes. Keeping this visual picture in mind can help you understand many different applications of de Bruijn graphs, including genome assembly (Section 3), transcriptome assembly (Section 6) and metagenome assembly (Section 6).
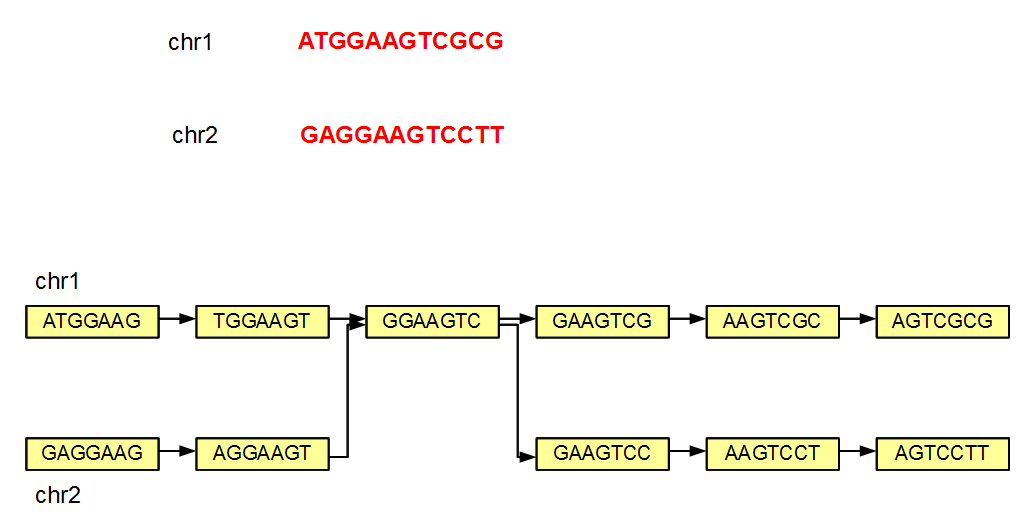
We note that even if a genome has multiple chromosomes, the de Bruijn graph of the genome may not remain disjoint. This point is best illustrated with a simple example (see figure below).