Note: These tutorials are incomplete. More complete versions are being made available for our members. Sign up for free.

De Bruijn Graphs for Metagenomic Samples
The knowledge derived about the microbial world through traditional sequencing had been minuscule, because it was nearly impossible to isolate every microbial species from the environment and sequence its genome. Next-generation sequencing opened up the field of metagenomics by allowing sequencing of an entire environmental sample without going through the step of isolating indiviual organisms. After sequencing of the combined sample, separation was done through computer algorithms. Researchers ventured to sequence microbial communities from all places including oceans around the world, deep inside mines, human gut, nostril and eyes, honey bee colonies, water under Antarctican ice, etc. In addition to satisfying intellectual curiosity about the invisible microbes living around us, metagenomic sequencing may help in medical diagnosis.
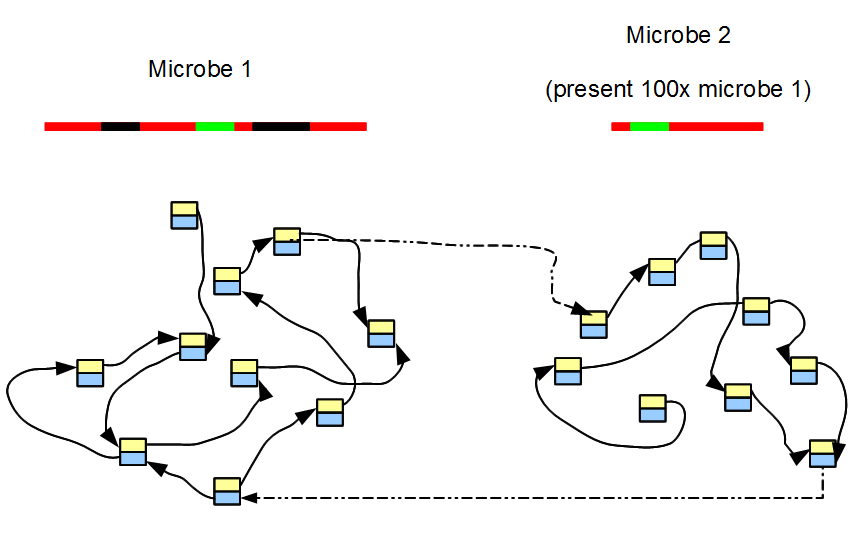
The field of metagenomics studies genetic materials collected from environmental samples. Each such sample typically contains hundreds or even thousands of microbes in different proportions. To visualize the structure of de Bruijn graph from a metagenomic sample, we follow the same conceptual steps as we did earlier for transcriptome and start with only two bacterial genomes. The de Bruijn graph for each bacterial genome can be constructed separately using the methods described in section 2.
De Bruijn graph of the combined sample merges the de Bruijn graphs of two bacterial genomes. If two microbes are evolutionarily distant, then the combined de Bruijn graph will be mostly disjoint except in the low-complexity regions. If two microbes are evolutionarily close, the combined de Bruijn graphs appears similar to the de Bruijn graph of a highly polymorphic diploid genome. The above procedure needs to be repeated tens or hundreds of times to conceptually visualize the de Bruijn graph of a large metagenomic sample.
While merging the de Bruijn graphs of different microbes, we need to keep in mind that the de Bruijn graph of a metagenomic sample also shows the characteristics of a transcriptome. Just like the genes in a transcriptome are present at various expression levels, chromosomes of different microbes are present in a metagenomic sample proportional to their relative abundance. However, individual graphs are more complex than those for genes, because genomes are larger than genes and contain many repetitive regions. Essentially metagenomic samples present all possible complexities (genome-like features, transcriptome-like features, high degree of polymorphism) together.